
A systematic study on the synthesis of α-Fe2O3 multi-shelled hollow spheres†
Abstract
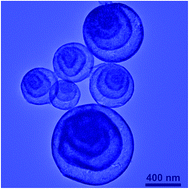
Hematite (α-Fe2O3) multi-shelled hollow spheres (MSHSs) have been prepared by a facile spray drying method using iron(III) citrate and sucrose as the precursors. The sucrose/iron(III) citrate ratio plays an important role in the morphology of the products. Well-defined MSHSs can be obtained in a wide sucrose/iron(III) citrate ratio of 0.25–1.5. The application of α-Fe2O3 MSHSs in lithium storage has been demonstrated. When used as the anode material for lithium-ion batteries, the α-Fe2O3 MSHSs exhibit a high reversible capacity of 979 mA h g−1, maintaining 861 mA h g−1 after 50 cycles.
 Please wait while we load your content...
Please wait while we load your content...

