
Functional and structural evidence for the catalytic role played by glutamate-47 residue in the mode of action of Mycobacterium tuberculosis cytidine deaminase†
Zilpa Adriana Sánchez-Quitianab,
Valnês Rodrigues-Juniorabc,
Jacqueline Gonçalves Rehma,
Paula Eichlera,
Daniela Barretto Barbosa Trivellad,
Cristiano Valim Bizarroab,
Luiz Augusto Basso*abc and
Diogenes Santiago Santos*ab
aCentro de Pesquisas em Biologia Molecular e Funcional (CPBMF), Instituto Nacional de Ciência e Tecnologia em Tuberculose (INCT-TB), Pontifícia Universidade Católica do Rio Grande do Sul (PUCRS), 6681/92A. Av. Ipiranga, 90619-900, Porto Alegre, RS, Brazil. E-mail: luiz.basso@pucrs.br; diogenes@pucrs.br; Fax: +55-51-33203629; Tel: +55-51-33203629
bPrograma de Pós-Graduação em Biologia Celular e Molecular, PUCRS, Porto Alegre, RS, Brazil
cPrograma de Pós-Graduação em Medicina e Ciências da Saúde, PUCRS, Porto Alegre, RS, Brazil
dLaboratório Nacional de Biociências, Centro Nacional de Pesquisa em Energia e Materiais, Rua Giuseppe Maximo Scolfaro 10000, 13083-970, Campinas, SP, Brazil
First published on 26th November 2014
Abstract
Strategies to combat tuberculosis (TB) are needed to kill drug-resistant strains and be effective against latent Mycobacterium tuberculosis, the causative agent of TB. Cytidine deaminase (CDA) catalyzes the hydrolytic deamination of cytidine to uridine, and belongs to the pyrimidine salvage pathway. The CDA from M. tuberculosis (MtCDA) is a target for the development of drugs against TB because it may be involved in latency mechanisms. The role of the conserved glutamate-47 (E47) residue was evaluated by construction of five mutant proteins (E47A, E47D, E47L, E47H, and E47Q). Mutants E47A and E47H were expressed in the insoluble fraction, whereas E47D, E47L and E47Q were soluble and purified. The E47D, E47L and E47Q mutants contained 1 mol of Zn2+ per mol of protein subunit. These mutations had no effect on the oligomerization state of MtCDA. Steady-state kinetic results showed that KM values for the E47D and E47Q mutants were not significantly altered, whereas there was a decrease in kcat values of 37-fold for E47D and 19-fold for E47Q mutant. No activity could be detected for E47L mutant. No kcat and kcat/KM dependence on pH values ranging from 4.0 to 11 were observed for E47D mutant from pH-rate profiles. A catalytic role was proposed for the γ-carboxyl group of E47, and its likely involvement in the stabilization of the transition state was suggested. Structural comparisons between E47D and E47Q mutants with the apo and holo forms of wild-type MtCDA reveal subtle differences that support this proposal.
Introduction
Tuberculosis (TB) continues to be a leading cause of death in the world due to a single infectious pathogen. It is estimated that one third of the entire human population is latently TB infected (LTBI). People with LTBI do not present symptoms of TB and are not infecting, but they are at risk of developing the active disease and becoming an infecting person. For latent and active TB, new drugs are urgently needed to shorten treatment and duration of therapy and target multiple drug-resistant strains, latent and nonreplicating bacilli.1,2Current antitubercular drugs have reduced or no effect against latent nonreplicating bacilli.3 This reduced effect may reflect the decreased activity of the various target enzymes under in vivo or nonreplicating conditions. Pyrimidine bases and nucleotides serve as nitrogen and carbon sources and are major energy carriers involved in nucleotide synthesis. There are two main routes for the synthesis of nucleotides: de novo and salvage pathways. The pathway for de novo synthesis of pyrimidine nucleotides leads to formation of uridine monophosphate (UMP) from simple precursors (ATP, HCO3−, H2O, glutamine). On the other hand, the pyrimidine salvage pathway reutilize nucleosides and pyrimidine bases derived from preformed nucleotides being preferentially utilized by bacteria under restricted energy availability, because it demands less energy than the de novo biosynthesis.4,5 Some metabolic pathways are presumed to be important for maintenance of viability under nonreplicating conditions.6 Accordingly, the pyrimidine salvage pathway enzymes are interesting targets due to low energy demand to synthesize nucleotides. Cytidine deaminase enzyme (CDA, cytidine/2′-deoxycytidine aminohydrolase; EC 3.5.4.5) catalyzes the deamination of cytidine/2′-deoxycytidine to form uridine/2′-deoxyuridine.7
The proposed mechanism of catalysis for Escherichia coli CDA includes a zinc atom in the active site that activates a water molecule to form a hydroxide ion and H+ by heterolytic bond cleavage.8,9 The proton and the hydroxyl anion add to, respectively, the N3 and the C4 atoms of the N3![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C4 double bond of cytidine to form a tetrahedral intermediate in the first step. In the second step, uridine is produced with the release of ammonia.8,9 Hydrogen bonding plays an important role in transition state stabilization for E. coli CDA, and the carboxymethyl group of Glu104 appears to minimize the activation barrier for deamination, not only by stabilizing the altered substrate in the transition state but also by destabilizing the enzyme–substrate and enzyme–product complexes.10–12
C4 double bond of cytidine to form a tetrahedral intermediate in the first step. In the second step, uridine is produced with the release of ammonia.8,9 Hydrogen bonding plays an important role in transition state stabilization for E. coli CDA, and the carboxymethyl group of Glu104 appears to minimize the activation barrier for deamination, not only by stabilizing the altered substrate in the transition state but also by destabilizing the enzyme–substrate and enzyme–product complexes.10–12
The cytidine deaminase superfamily includes enzymes that act on the in situ deamination of bases in both RNA and DNA, which are involved in gene diversity and in anti-virus defense, and enzymes that catalyze reactions of free nucleosides, nucleotides or bases such as cytidine deaminases (CDD/CDA), deoxycytidylate monophosphate deaminases (dCMP), and guanine deaminase (GuaD). These enzymes are primarily involved in the salvage of pyrimidines and purines, or in their catabolism in bacteria, eukaryotes and phages.13,14 The mononucleotide family includes a homodimeric form (D-CDA) of cytidine deaminase, such as E. coli and Arabidopsis thaliana15,16 and a related homotetrameric form (T-CDA) with a much smaller subunit, which is found in most mammalians, such as human, and bacteria such as Bacillus subtilis and Mycobacterium tuberculosis.17–19 D-CDA consists of two symmetrical subunits, each monomer containing three domains: a small N-terminal domain, a catalytic domain containing a zinc atom, and a C-terminal domain, that contains a cavity described as “broken active site”.9 T-CDA consists of four identical monomers with one active site per monomer. Each monomer has a molecular weight of 14.9 kDa for human CDA and B. subtilis, and 13.9 kDa for M. tuberculosis CDA (MtCDA), and is structurally similar to the catalytic domain of the E. coli D-CDA.9 D-CDA and T-CDA have a zinc atom in the active site.8
The homotetrameric MtCDA is composed of four identical subunits of 13.9 kDa with one active site per subunit.19 Each monomer has five β-strands and five α-helices. Secondary structural elements are arranged in a three-layer core α–β–α domain with antiparalell β-sheet composed by the five strands, the last two strands of this structure form a β-wing flexible region. Structural studies of MtCDA in complex with products (uridine and deoxyuridine) show three main regions related to product binding pocket.20 Region 1 and region 2 are located in the tetramer interface and are, therefore, also involved in quaternary structure stabilization. Region 1 comprises residues V22 to F27, which are not involved in ligand binding. Region 2 is comprised of residues T42 to C56, which contribute to the stability of ligands in the entrance of the binding cavity. The third region, which is comprised of C-terminal residues (V110–F123), is located close to an adjacent subunit and π–π stacking interactions between the pyrimidine moiety of products (uridine and 2′-deoxyuridine) and F123 appears to contribute to ligand binding.20 The human CDA region involved in substrate binding contains the residues 32PYSHF36 and 56ENACYP61, similar to those described for B. subtilis CDA (20PYSKF27, 44ENAAYS49) and for MtCDA (23PYSRF27, 47ENVSYG52).18,19,21 Conserved residues in tetrameric CDA enzyme involved in catalysis are 65CAERTA70 for human CDA, 53CAERTA58 for B. subtilis CDA, and 56CAECAV61 for MtCDA.18–21
In previous pH-rate profile studies of MtCDA, protonation of a single ionizable group with an apparent pKa value of 4.3 (±1) reduced the catalytic activity and protonation of a group with apparent pKa of 4.7 (±0.7) appeared to be involved in cytidine binding.19 The γ-carboxyl groups of E47 and E58 were proposed to be involved in substrate binding and/or catalysis.19 Glutamic acid residues have been shown to be conserved in D-CDAs and T-CDAs and were implicated in substrate binding.15
The goal of this study was to evaluate the role, if any, of MtCDA E47 in substrate binding and/or catalysis, protein oligomerization and coordination of zinc atom. In order to test these predictions, we employed site-directed mutagenesis methodology to produce five mutants for the same residue. Functional and structural characterization of these mutants here described prompted us to propose a role played by E47 residue in the catalytic mechanism of MtCDA enzyme.
Results and discussion
Amplification and cloning
MtCDA coding sequence was obtained from M. tuberculosis H37Rv genomic DNA. Two separate PCR reactions were carried out, and two fragments of a target sequence were amplified by using, for each reaction, one flanking and one mutagenic primer. The two intermediate products with complementary terminal sequences form a new template DNA by duplexing in a second reaction or overlap extension-PCR, in which the two fragments were joined using two outmost flanking primers. Fig. S1† shows the product from two PCR reactions for mutated gene. Cloning of the mutated PCR products was performed using the CATATG and AAGCTT restriction sites in the pET-23a (+) expression vector. E. coli DH10B electro-competent cells were transformed and recombinant clones were identified by restriction enzyme digestion. Mutagenesis was confirmed by nucleotide sequencing and all recombinant clones contained the expected mutated codons, E47A (GAA → GCG), E47D (GAA → GAT), E47H (GAA → CAC), E47L (GAA → CTG), and E47Q (GAA → CAG).Expression of recombinant proteins
Each recombinant plasmid was purified and transformed into E. coli BL21 (DE3) as described for wild-type MtCDA.19 Recombinant protein expression was analyzed by SDS-PAGE. Two (E47D and E47Q) of five mutant proteins were expressed in the soluble fraction (SF) and exhibited the expected molecular mass of ∼13.9 kDa and were expressed at similar levels to the wild-type protein (Fig. S2†). One recombinant protein (E47L) showed low levels of expression in soluble form, and required the use of low molecular weight compounds (chemical chaperones) in the culture medium to, purportedly, stabilize proteins in their native conformations, as described under materials and methods section. The results indicated that the presence of DMSO 6% in the TB culture medium increased the expression of the E47L mutant MtCDA protein (Fig. S2†).Two constructs failed to express the expected recombinant proteins (E47A and E47H) in the soluble fraction. There are a number of factors that could influence and impair folding leading to formation of inclusion bodies.22 Expression of these constructs was evaluated in alternative E. coli strains (BL21 (NH), C41, Rosetta, Rosetta-gami), at lower temperature of cultivation (30 °C), and three different growth media (LB, TB and 2YT) and both supernatants and pellets of cell lysates were screened for the presence of recombinant proteins. Unfortunately, E47A and E47H were expressed in insoluble form for all host cells and conditions tested (data not shown). Although inclusion body formation can greatly simplify protein purification, there is no guarantee that the in vitro refolding will yield large amounts of biologically active product. Moreover, inclusion body purification schemes present a number of problems such as: use of denaturants that can cause irreversible modifications of protein structure that will elude all of the most sophisticated analytical tests, refolding usually must be done in very dilute solution and the protein reconcentrated, and refolding encourages protein isomerization, leading to precipitation during storage.23 Since one of the goals of the present work is to assess the role of E47 residue in MtCDA-catalyzed chemical reaction, we deemed appropriate not to try unfolding and refolding protocols.
Purification of soluble mutant proteins
Purification of the recombinant E47D and E47Q mutants was performed according to the methodology described in materials and methods section. The eluted fractions, after three chromatographic steps, were analyzed. According to analysis on SDS-PAGE, the purification protocol yielded 9.1 mg and 4.5 mg of, respectively, E47D and E47Q homogeneous soluble protein from 10 g of wet cells. The same purification protocol yielded 35 mg of homogeneous soluble wild-type MtCDA from 10 g of wet cells.19 Therefore, the yields for E47D and E47Q mutant proteins were, respectively, 74% and 87% lower than the yield obtained for wild-type MtCDA. A number of factors can interfere with protein yield. For instance, a lower expression level of M. tuberculosis CDA mutants can be invoked to explain the lower yield of homogeneous proteins as compared to wild-type MtCDA even though the same purification protocol was employed to purify the mutant and wild-type proteins. To avoid cross contamination between the different mutant enzymes it was necessary to use a set of columns for each protein.The E47L recombinant protein was expressed in low levels and its purification was not successful using the same protocol. An alternative purification protocol was thus developed and is described in the Materials and methods section. The purification protocol employed to purify E47L mutant protein was also employed to purify wild-type MtCDA, yielding soluble wild-type MtCDA with specific activity values of 4.75 U mg−1 for cytidine and 3.46 U mg−1 for 2′-deoxycytidine. These values are comparable the values for wild-type MtCDA purified via the original protocol (5.44 U mg−1 for cytidine and 3.75 U mg−1 for 2′-deoxycytidine) previously reported,19 suggesting that the protocol employed for the purification of E47L mutant cannot be invoked to explain its lack of enzyme activity. The E47L was obtained partially homogeneous in solution according to analysis by 15% SDS-PAGE. The purification yielded 1.4 mg of E47L from 4 g of wet cells. Mutant proteins were stored at −80 °C. Fig. S3† shows the results from SDS-PAGE analysis of three recombinant mutant proteins purified by chromatography.
Molecular mass determination by mass spectrometry (MS)
In order to determine the wild-type MtCDA, E47D, E47L, and E47Q molecular masses, intact protein analysis was performed using an Orbitrap analyzer (see Experimental procedures). The average spectra from 100 scans for wild-type MtCDA and E47D proteins, 1000 scans for E47L, and 1238 scans for E47Q with charge states spanning from 7 + to 18+ were detected with isotopic resolution (Fig. S4–S7†).The high-resolution FTMS spectra contained isotopic envelopes for each charge state (see Fig. S4B† for the isotopic envelope of charge state 14+ of wild-type MtCDA as an example) which allowed us to obtain the monoisotopic molecular mass for each CDA form. In Table 1 we show the theoretical (N-terminal methionine removed) and experimentally obtained molecular masses for wild-type and mutant MtCDA forms, differences in mass between expected and experimental measurements, and also the parts-per-million (ppm) accuracy of measurements based on the expected theoretical values.
| Protein | Theoretical monoisotopic mass (Da) | Experimental monoisotopic mass (Da) | Δtheor − exp (Da) | Accuracy (ppm) |
|---|---|---|---|---|
| a With a disulfide bond formed.b With two disulfide bonds formed. | ||||
| wtCDA | 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 931.9575 931.9575 |
13931.9586 |
0.0011 | 0.0789 |
| E47D | 13917.9419 |
13917.9684 |
0.0265 | 1.9 |
| E47Q | 13930.9735 |
13928.9716 |
2.0019 | 143.70 |
| E47Qa | 13928.9579 |
13928.9716 |
0.0137 | 0.98 |
| E47L | 13915.9990 |
13911.9993 |
3.9997 | 287.42 |
| E47Lb | 13911.9677 |
13911.9993 |
0.0316 | 2.3 |
Our data strongly indicate that the N-terminal methionine was removed in all CDA forms studied. Moreover, the molecular mass of wild-type MtCDA was determined with sub-ppm accuracy (0.079 ppm–0.0011 Da) while we obtained a difference of 1.9 ppm (0.0265 Da) between expected and experimentally obtained monoisotopic masses for E47D MtCDA mutant. However, the accuracies in measurements for both E47Q and E47L mutants were rather low (143.70 ppm and 287.42 ppm, respectively). If we consider the existence of disulfide bonds between cysteine residues (one disulfide bond for E47Q mutant and two for E47L mutant), the correspondence between expected and obtained values greatly improves, reaching sub-ppm levels for E47Q (0.98 ppm–0.0137 Da). Therefore, our MS data suggest that E47Q mutant is likely to contain one disulfide bond (−2.0156 Da) whereas E47L mutant is likely to contain two disulfide bonds (−4.0313 Da).
Protein identification by LC-MS/MS peptide mapping experiments
LC-MS/MS peptide mapping experiments were performed and the MS/MS spectra of wild-type and mutant proteins were compared with the M. tuberculosis proteome including the wild-type form of MtCDA and the mutants. Peptides obtained covered the wild-type and the mutated sequences in 100% for wild-type MtCDA and E47D mutant, 92% and 74% for E47Q and E47L mutants, respectively.Determination of oligomeric state of mutant proteins
To establish whether the mutant proteins have the same oligomeric state of wild-type MtCDA, N-PAGE was performed. Under nondenaturing conditions, in which protein activity, native charge and conformation are maintained, electrophoretic separation showed that E47D and E47L mutant proteins have an apparent molecular mass of approximately 55 kDa (Fig. S8†). This result suggests a homotetrameric state for these MtCDA mutants as the subunit molecular mass value is 13.9 kDa. This is in agreement with the oligomeric state of wild-type MtCDA.19A lower migration rate was observed for E47Q as compared to E47D and E47L mutants (Fig. S8†). Interestingly, the E47Q mutant is prone to precipitation at larger protein concentrations (observed in crystallization trials) and is less stable than wild-type MtCDA. As mass spectrometry showed that the E47Q protein has the expected subunit molecular mass, this mutation could cause alterations on protein conformational states.
Metal analysis by ICP-OES
Determination of metal concentration and identity by ICP-OES yielded the following results for Zn2+ concentrations: E47D = 7.3 ± 0.3 mg L−1, E47L = 1.8 ± 0.2 mg L−1, and E47Q = 8.2 ± 0.6 mg L−1. These results indicate the presence of one mol of Zn2+ (E47D 111 μM, E47L 27 μM and E47Q 125 μM) per mol of enzyme subunit (143 μM, 35 μM and 143 μM respectively), as observed for wild-type MtCDA.19 These findings suggest that E47 residue plays no role in coordinating the zinc cation in MtCDA active site. These results are not surprising as structural studies of MtCDA revealed that E47 residue is involved in the ribose binding loop and is located away from the zinc atom.19 The thiolate side chain of Cys56, Cys89, and Cys92 and the carboxylate side chain of Glu58 residue have been implicated in the coordination of zinc atom for MtCDA,19 in agreement with the pattern reported for B. subtilis CDA.15Kinetics properties
The effect of substitutions in the E47 position on the steady-state kinetics parameters for MtCDA enzyme was determined using both a continuous spectrophotometric assay and a discontinuous HPLC method. Enzyme velocity measurements for E47D mutant was performed as previously reported for wild-type MtCDA using cytidine as substrate.19 For E47Q mutant MtCDA, reverse-phase HPLC was used to monitor the slow conversion of cytidine to uridine as described in Experimental procedures section. The results for the E47D mutant (Fig. 1) were fitted to eqn (1). The results for the E47Q mutant were first fitted to a linear equation (Fig. 2A) to obtain initial velocity values (v0), and the latter were plotted as a function of increasing cytidine concentration (Fig. 2B). The hyperbolic increase in v0 as a function of cytidine concentration was fitted to eqn (1) to determine KM and Vmax (Fig. 2B). The values of kcat for E47D and E47Q mutants were determined from eqn (2). The KM values for the E47D and E47Q mutants were not significantly altered compared to wild-type MtCDA (Table 2). On the other hand, there was a 37-fold decrease in the kcat value for the E47D mutant, and a 19-fold decrease in kcat for the E47Q mutant (Table 2). | ||
| Fig. 1 Determination of steady-state kinetic parameters for E47D mutant. The data for the hyperbolic cytidine saturation curve were fitted to eqn (1). | ||
 | ||
| Fig. 2 Determination of kinetic parameters for E47Q mutant. (A) Product (uridine) formation versus time of substrate (cytidine) enzymatic hydrolysis at increasing concentrations estimated by HPLC. (B) Initial velocity, v0, plotted against increasing substrate concentrations for a reaction obeying the Michaelis–Menten kinetics. The data were fitted to eqn (1). | ||
The results for the E47D mutant demonstrate that shortening the carbon chain whereas preserving the carboxylic group impairs catalysis. For the E47Q mutant, the non-ionizable amide side chain of glutamine eliminates the negative charge of the side chain whereas keeping its ability to form hydrogen bonds. Although the Michaelis–Menten constant (KM) is not a true dissociation constant; it can be regarded as an apparent dissociation constant that may be treated as the overall dissociation constant of all enzyme-bound species.24 The catalytic constant (kcat) is a first-order rate constant that refers to the properties and reactions of the enzyme–substrate, enzyme–intermediate, and enzyme–product complexes, including the enzyme-catalyzed chemical reaction.24 Accordingly, the results for the E47D and E47Q mutants provide experimental evidence for the catalytic role played by the negatively charged carboxyl group (likely electrostatic catalysis) and the importance of carbon chain length in the mode of action of MtCDA. The replacement of E47 with leucine produced an inactive mutant protein, as no activity could be detected using either a continuous spectrophotometric assay or a discontinuous HPLC method. Although the side chain of leucine has the same volume of the R group of glutamate, its aliphatic isobutyl chain cannot form hydrogen bonds and is not charged. As previously shown for the three-dimensional structure of wild-type MtCDA in complex with 2′-deoxyuridine,20 the E47 residue is located in the active site but does not interact directly with the zinc atom, as shown for other organisms.9,18,21 It, however, forms an important hydrogen bond to 3′-OH group of the pentose of 2′-deoxyuridine.21 As the E47D, E47L, and E47Q mutant enzymes are likely homotetrameric and have a zinc atom bound per subunit, the differences in steady-state parameters as compared to wild type MtCDA are thus due to the catalytic role of E47. These results are in agreement with the pH-rate profiles previously reported for MtCDA.19 The corresponding E91 in E. coli CDA has been shown to form an H-bond with the 3′-OH group of substituent ribose, a substrate moiety that is not directly involved in chemical reaction.9 Site-directed mutagenesis studies for the E91A mutant of E. coli CDA showed a 500-fold increase in KM and a 32-fold reduction in kcat using cytidine as substrate.25 These results prompted the proposal that the E91 residue in E. coli CDA (corresponding to E47 in MtCDA) plays a role in transition state stabilization.25 It should be pointed out that, as assumed by Richard Wolfenden and colleagues for E. coli CDA,10 we have considered kcat to represent the rate constant for chemical transformation of the substrate at the enzyme's active site and KM to describe the dissociation constant of MtCDA:substrate complex. No change in KM values were observed for E47D and E47Q MtCDA mutant enzymes, contrary to the results observed for the E91A E. coli CDA.25 It thus appears that the E47 residue in MtCDA plays a role in catalysis and a minimal, if any, role in substrate binding. These results are in agreement with the pH-rate profiles previously reported for MtCDA.19 Interestingly, glutamate residues have been shown to be implicated in electrostatic stabilization of intermediate(s) or transition state and shown to play a prominent role in proton shuttling in the mode of action of a number of enzymes.26,27
pH profile
The pH dependence of kcat and kcat/KM for cytidine was studied only for the E47D mutant MtCDA, as this mutant permits collecting kinetic parameters using a continuous spectrophotometric assay. As previously reported for wild type MtCDA enzyme,19 the pH-rate profile for kcat suggested that protonation of a group with an apparent pKa value of 4.3 abolished enzyme activity, and pH-rate profile for kcat/KM showed that protonation of a single ionizable group with an apparent pKa value of 4.7 decreased cytidine binding. The pH-rate profile of the kinetics parameters for E47D mutant using cytidine as substrate is shown in Fig. 3. While wild type MtCDA had the broadest pH range of activity and showed a decrease at low pH,19 the mutant form (E47D) result in a flat pH profile with significant drop in the catalytic rate. The shapes of the pH-activity profiles of the mutant enzyme were altered, suggesting that removal of the general acid through site-directed mutagenesis results in decrease of catalytic activity. These results suggest that unprotonated E47 is required for catalysis. However, how can a residue at 10.6 Å from the C4 of the base, where the nucleophilic attack occurs, affect the catalysis? Possibly, the aspartate side chain of E47D cannot keep the ribose moiety in place to assist the correct positioning of the pyrimidine ring of cytidine, thereby impairing either proton transfer to N3, or proper positioning of zinc-bound hydroxyl group to attack the C4 of cytidine. Interestingly, the E91 residue of E. coli CDA (corresponding to E47 of MtCDA) was shown to play a role in stabilization of the transition state for the deamination reaction.25 As mentioned above, no change in KM value was observed for E47D MtCDA mutant, whereas a 500-fold increase in KM was observed for E91A E. coli CDA mutant.25 | ||
| Fig. 3 The pH-profile of E47D MtCDA protein. pH-rate profiles for E47D MtCDA catalyzed reaction. (A) pH dependence of logkcat; (B) pH dependence of logkcat/KM. | ||
Crystal structures of E47D and E47Q mutant MtCDA
In order to evaluate the effect of E47 substitutions on the structure of MtCDA active site, the crystal structures of apo E47D and apoE47Q MtCDA mutants were solved (Fig. 4, Table S1†). Superposition of the apoE47 mutants with the wild type apo MtCDA and holo MtCDA structures revealed subtle differences that can assist in pointing out the role of E47 residue in the CDA catalytic mechanism (Fig. 4). The structure of E47Q mutant revealed that the glutamine side chain is accommodated in a very similar position to the glutamate counterpart in the wild-type MtCDA. However, the best fitted glutamine rotamer (rotamer 1) points the Nε atom to the binding site; whereas the Oε atom of wild-type MtCDA interacts with the main-chain N atoms of Y24 and S25 (Fig. 4, right panel). The two possible glutamine rotamers – rotamer 1 mentioned above and rotamer 2 (representing the side chain conformation in which the Oε atom is pointed to the binding site) – were tested and refined. Rotamer 1 (shown in Fig. 4) was chosen based on B-factor analysis (Table S2†). The chemical groups of Q47 that interact with substrate and product depend on the rotamer being considered. For rotamer 1 (Fig. 4, right panel) the amide nitrogen is pointing towards the ribose moiety of uridine, whereas the negatively charged carboxylic group of E47 would interact with the 3′-OH group of uridine for wild-type MtCDA. As a decrease in kcat and no change in KM were observed for E47Q MtCDA mutant (Table 2), the functional and structural data suggest that the side chain of E47 residue plays a role solely in catalysis (e.g., transition state stabilization). The hydrogen bond interaction between Q47Nε and the substrate is still possible; however, the geometry and strength of the Q47Nε interaction with ligands can result in improper positioning of the substrate and reaction intermediates during catalysis. This can lead to changes in kcat not necessarily affecting the KM parameter. No significant changes were observed in the bottom of the E47Q catalytic site (Fig. 4, right panel), with exception of a little disorder in the electron density around the zinc atom and the C56 and C92 side chains. No clear evidences of S–S bonds were observed in the MtCDA E47Q crystal structure. | ||
| Fig. 4 Apo MtCDA, E47D and E47Q binding sites. The E47D tetramer is shown in the middle panel, while the binding sites of apoE47D (left panel) and apoE47Q (right panel) are zoomed. Important residues of binding sites were superposed with the holo-wild-type MtCDA structure biding uridine (PDB ID 3LQP). Oxygen, nitrogen and sulphur atoms are shown in red, blue and yellow, respectively. Carbon atoms are pink for apoE47Q (right panel), light blue for apoE47Q (left panel) or grey for holo wild-type MtCDA (wtCDA:uridine). Amino acid residues are labelled and interatomic distances are given in Angstroms. | ||
The MtCDA E47D mutant crystal structure points to local perturbations in the active site (Fig. 4, left panel). Although the E47D substitution does not change the chemical nature of ligand interaction, shortening the side chain at position 47 affects ligand position at the catalytic center. The distance between the carboxyl side chain oxygen of D47 and the 3′-OH of the sugar moiety of uridine increased from 2.4 Å in the wild-type MtCDA to 3.3 Å in the E47D mutant (Fig. 4, left panel). As a decrease in kcat and no change in KM were observed for E47D MtCDA mutant (Table 2), these functional and structural data provide further support for a catalytic role played by E47 in MtCDA. Unfortunately, efforts to obtain the crystal structures of the E47 mutants in the presence of ligands were unsuccessful. However the apo mutant structures presented here can at least shed light on this proposal.
Conclusions
Production of mutant enzymes in which the conserved residue, E47 was replaced with aspartate, leucine, and glutamine residues could be accomplished. No activity could be measured for the E47L mutant. The E47D and E47Q mutations resulted in decreased kcat and no effect on KM for cytidine was observed. Accordingly, a catalytic role was proposed for the E47 side chain in MtCDA-catalyzed hydrolytic deamination. In addition, the results showed that the E47 residue is not involved in zinc ion coordination nor in oligomerization state or MtCDA. Interestingly, although the residues E47 and/or E58 residues were proposed to play a role in catalysis of and/or substrate binding to MtCDA,19 the results here presented show that E47 plays a minimal role, if any, in substrate binding. Interestingly, crystal structure data for MtCDA in complex with product showed that the E47 side chain interacts with the ribose moiety of uridine.20 This result would suggest a role for E47 in substrate/product binding, which is not borne out by the data here presented. At any rate, site-directed mutagenesis of the conserved E58 residue in MtCDA19 will have to be pursued to evaluate whether or not this residue plays any role as it appears to be more appropriately positioned for catalysis. It is hoped that the results here reported contribute to an increased understanding of the functional roles of amino acid side chains in enzyme catalysis, and also show that a functional role for an amino acid residue in the mode of action of a particular enzyme has to be demonstrated by biochemical and, if possible, structural data.Experimental procedures
Residue selection and primer design
pH-rate profile studies and multiple sequence alignment suggested that the conserved amino acid Glutamate in the position 47 (E47) from MtCDA is involved in catalysis and/or substrate binding.19 Site-directed mutagenesis was thus carried out, and E47 was replaced with either alanine (E47A), aspartate (E47D), histidine (E47H), leucine (E47L), and glutamine (E47Q).Primers were designed with the aid of Primer3Plus and were based on the CDA M. tuberculosis H37Rv genome sequence. The mutagenesis procedure required four oligonucleotides: two flanking primers, which were positioned upstream (A) and downstream (D) of the mutation site; and two mutagenic primers, Forward mutagenic (C) and, Reverse mutagenic (B) with at least a 15 bp overlap between adjacent fragments. The mutation site was located in the middle of the mutagenic primers. Forward and reverse flanking primers containing restriction sites for NdeI (CATATG) and HindIII (AAGCTT) for cloning into PCR-Blunt vector and subcloning into pET23a (+) expression vector were also designed. Primers used in the polymerase chain reaction (PCR) are given in Table 3.
| Primers | Sequence (5′ to 3′)a | Length of overlapping sequence (bases) |
|---|---|---|
| a Nucleotide substitution is showed in boldface and lower case; Underlined nucleotides represent cloning site; CATATG and AAGCTT restriction sites for NdeI and HindIII enzymes, respectively. | ||
| MTBcddFor_U | GC![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) ![[A with combining low line]](https://www.rsc.org/images/entities/char_0041_0332.gif) ![[T with combining low line]](https://www.rsc.org/images/entities/char_0054_0332.gif) ![[G with combining low line]](https://www.rsc.org/images/entities/char_0047_0332.gif) CCTGATGTCGATTGGAATATGCTG CCTGATGTCGATTGGAATATGCTG |
|
| MTBcddRev_D | GATCACCGGCGTTCCCGGGGGAG |
|
| MTBcddE47A FM | GTGACCGGATGCAACGTGgcgAACGTCTCGTATGGCTTG | 18 |
| MTBcddE47A RM | CAAGCCATACGAGACGTTcgcCACGTTGCATCCGGTCAC | |
| MTBcddE47D FM | CCGGATGCAACGTGgatAACGTCTCGTATGGC | 14 |
| MTBcddE47D RM | GCCATACGAGACGTTatcCACGTTGCATCCGG | |
| MTBcddE47H FM | CCGGATGCAACGTGcacAACGTCTCGTATGGC | 14 |
| MTBcddE47H RM | GCCATACGAGACGTTgtgCACGTTGCATCCGG | |
| MTBcddE47L FM | GTGACCGGATGCAACGTGctgAACGTCTCGTATGGCTTG | 18 |
| MTBcddE47L RM | CAAGCCATACGAGACGTTcagCACGTTGCATCCGGTCAC | |
| MTBcddE47Q FM | GTGACCGGATGCAACGTGcagAACGTCTCGTATGGCTTG | 18 |
| MTBcddE47Q RM | CAAGCCATACGAGACGTTctgCACGTTGCATCCGGTCAC | |
Site-directed mutagenesis method
Site-directed mutagenesis was carried out using a two-step PCR procedure to replace the GAA codon using an overlap extension-PCR method (OE-PCR) (Fig. 5).28 In short, in the first step; two simultaneous PCR reactions were performed. One reaction was performed with a primer pair that included the A primer and the B primer; the other reaction contained the D primer and the C primer. The PCR reaction were carried out using 50 ng of M. tuberculosis H37Rv genomic DNA, 0.2 mM dNTPs, 10 pmol of each primer, 2.5 U of PFU DNA polymerase and 1× reaction buffer in a 50 μL reaction volume. For amplifications, an MJ Research PTC-200 (Peltier Thermal Cycler) was used with the following parameters: 5 min at 98 °C followed by 30 cycles of 45 s at 95 °C, 45 s at 60 °C and 45 s at 72 °C and a final extension of 10 min at 72 °C. PCR products were analyzed by 1% agarose gel electrophoresis and gel band was purified using the QIAquick Gel Extraction kit according to the manufacturer's instruction (QIAGEN). The second PCR was performed to obtain full-length mutated fragments; the two products were mixed and used as templates, with the A and D primers. The reaction was performed using the same conditions as the first round of PCR amplification. This overlap extension-PCR yielded a full-length DNA fragment, which was gel purified before cloning.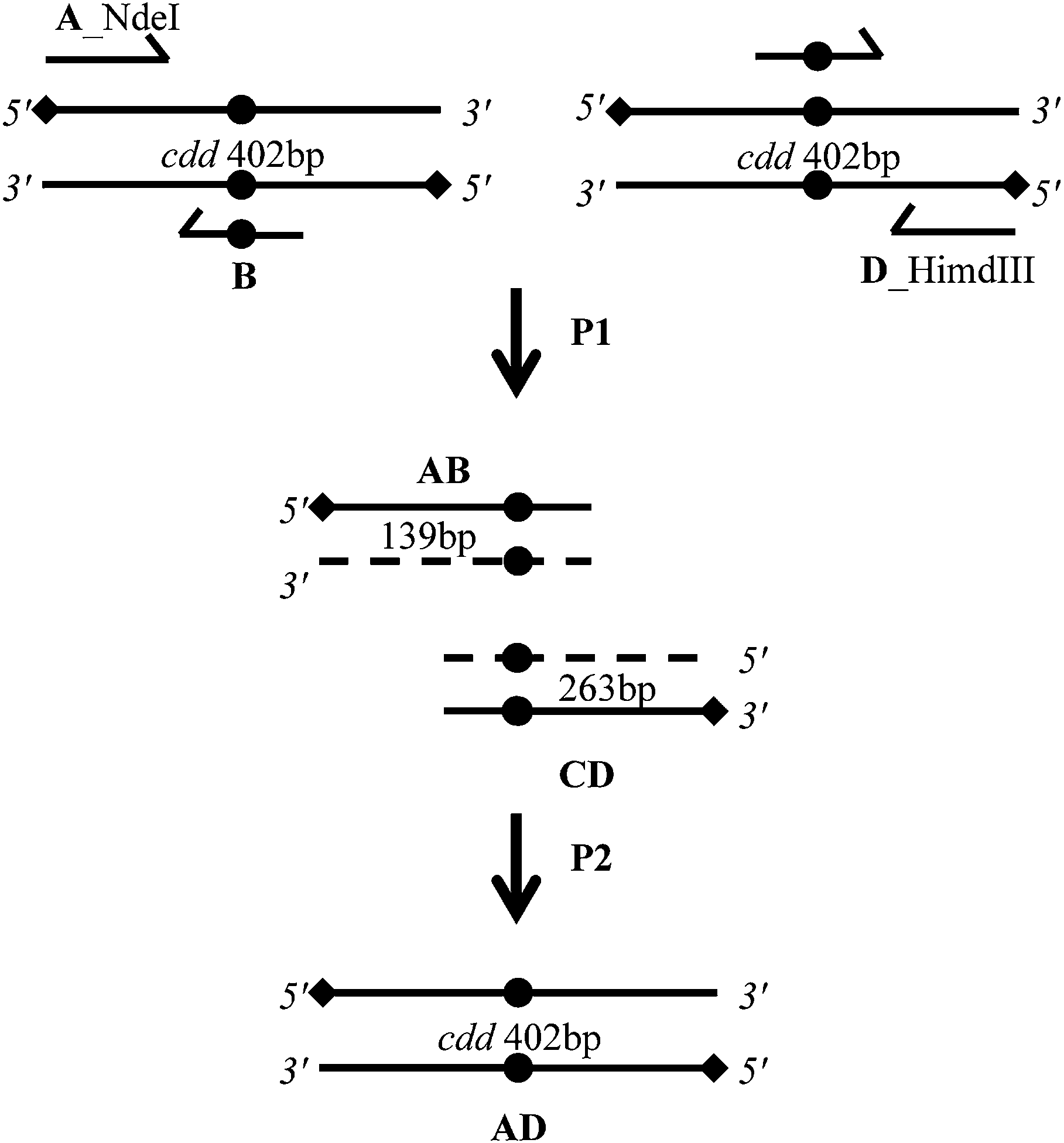 | ||
| Fig. 5 Schematic diagram of the overlap extension polymerase chain reaction (OE-PCR) method for site-directed mutagenesis. A nucleotide substitution (solid circle) was introduced into a template DNA (cdd gene) through two different steps of PCR reactions (P1 and P2). The first step (P1) was performed in a separate PCR reaction, two fragments of a target sequence (solid circle) was amplified by using forward flanking primers (A) and reverse mutagenic primers (B) for one reaction, and reverse flanking primers (D) and forward mutagenic primers (C), resulting in an amplification of the intermediate products AB and CA. Owing to their terminal complementarity, these products form a new template DNA by overlapping. Subsequently extension occurs in a second step (P2) with the help of two flanking primers (A and D). Finally, the recombinant PCR product can be cloned into NdeI and HindIII restriction sites. | ||
Cloning and sequencing of mutated genes
The full-length mutated PCR product was ligated into the pCR-Blunt cloning vector and subcloned into the pET-23a (+) expression vector (Novagen) using T4 DNA ligase. E. coli DH10B (Novagen) electro-competent cell were transformed and recombinant clones were identified by digestion with NdeI and HindIII. The sequences of the mutants of cdd gene were verified to both confirm the insertion of the desired mutation and to ensure that no unwanted mutations were introduced by the PCR steps, using the ABI-Prism 3100 Genetic Analyzer (Applied Biosystems).Expression of recombinant proteins
After confirming the presence of the mutated codon in the five substitutions (E47A, E47D, E47H, E47L and E47Q), the recombinant plasmids were transformed into E. coli BL21 (DE3) competent cells, and cells grown in LB medium for 6 h at 37 °C and 180 rpm after cell cultures having reached an OD value of 0.4–0.6, in the absence or IPTG (isopropyl-β-D-thio-galactoside) induction, following the protocol previously reported for wild type MtCDA.19 Since the E47L mutant enzyme was a partially soluble protein it was necessary to use chemical chaperones to increase the solubility of this mutant. Cultures (50 mL) of E. coli BL21 (DE3) harboring pET23a (+)::E47L were grown at 37 °C in terrific broth (TB), supplemented with 100 μg mL−1 ampicillin, in presence of a chemical chaperone (dimethyl sulphoxide, DMSO 6.0%). The negative control consisted of the same culture described above in the absence of DMSO 6.0%. Bacterial cultures were grown for 6 h at 37 °C and 180 rpm to an optical density (OD600 nm) of 0.4–0.6. Cells were harvested by centrifugation at 24400× g for 30 min, resuspended in buffer A (50 mM Tris–HCl pH 7.5), and lysed by sonic disruption.
Purification of MtCDA mutants
Cultures (500 mL) of E. coli BL21 (DE3) harboring the mutant plasmids were grown at 37 °C in LB broth, supplemented with 100 μg mL−1 ampicillin. After 6 h of vigorous shaking at 37 °C, cells were harvested by centrifugation at 24400× g for 30 min, resuspended in buffer A (50 mM Tris–HCl pH 7.5), and lysed by sonic disruption. After centrifugation at 24400× g for 30 min, the supernatant was loaded on a QFF (column volume 47 mL) anionic exchange chromatography column equilibrated with buffer A and eluted by a linear gradient of buffer B (50 mM Tris–HCl pH 7.5, 1 M NaCl). Fractions containing the enzyme were pooled and concentrated by ultrafiltration on PM-3 membrane (Spectrum LAB) and loaded onto a gel filtration Sephacryl 200 HR 26/60 equilibrated with buffer A. The mutant MtCDA fractions eluted from gel filtration were incubated with ammonium sulphate to a final concentration of 1 M and loaded on a Butyl Sepharose High Performance Hiload 16/10 (GE Healthcare) equilibrated with buffer C (50 mM Tris–HCl pH 7.5, 1 M ammonium sulphate). The column was washed with seven column volumes of buffer C and the adsorbed proteins were fractionated with a 20 column volume linear gradient from 1 to 0 M ammonium sulphate. The fractions obtained from the hydrophobic interaction chromatography were pooled and stored at −80 °C. Since the E47L mutant could not be purified using the protocol describe above, a different purification protocol had to be established. The supernatant, obtained by centrifugation 24400× g for 30 min, was treated with 1.5 M ammonium sulfate and the resulting precipitate was suspended in 9 mL of buffer A (crude extract). The crude extract was loaded on a Superdex 200 size exclusion column (GE Healthcare) previously equilibrated with buffer A. Fractions containing mutant E47L were pooled, loaded on DEAE CL6B anionic exchange chromatography equilibrated with buffer A, and eluted by a linear gradient of buffer B. The fractions containing homogeneous enzyme were pooled, dialyzed against 20 mM Tris–HCl pH 7.5 buffer, concentrated, and stored at −80 °C. A High performance liquid chromatography (HPLC) system (ÄktaPurifier) was employed for protein purification, and all purification steps were performed at 4 °C. Protein fractions were analyzed by 15% SDS-PAGE stained with Coomassie Brilliant Blue.29 Protein concentration was determined by the method of Bradford using the BioRad protein assay kit (Bio-Rad) and bovine serum albumin as standard,30 and confirmed by absorbance spectroscopy31 using a calculated molar absorption coefficient (ε) value of 9750 M−1 cm−1.
Molecular mass determination by mass spectrometry (MS)
Intact protein analysis was performed by direct injection of samples (reconstituted in acetonitrile 50%: water 49%: formic acid 1%) into an IonMax electrospray ion source. We applied 4.5 kV in positive ion mode, 250 °C of capillary temperature, 48 V of capillary and 170 V of tube lens voltage. Spectra with isotopic resolution (600–2000 m/z range for wild-type MtCDA and E47D mutant or a 800–1600 m/z range for E47Q and E47L mutants) were collected in FTMS mode using a Thermo Orbitrap Discovery XL (Thermo Electron Corp., San Jose, CA) at a nominal resolution of 30000 at 400 m/z. Charge state deconvolutions of averaged data from 100 to 1238 spectra were performed, depending on the protein sample, using the software Xtract (Thermo Electron Corp., San Jose, CA).
Protein identification by LC-MS/MS peptide mapping experiments
Purified MtCDA samples (1 nmol) of wild-type and E47D, E47Q and E47L MtCDAs were trypsin digested using a protocol adapted from Klammer & MacCoss.32 The resulting peptide mixtures were subjected to nanochromatography (nanoLC Ultra 1D plus – Eksigent, USA) using a packed in-home capillary column33 (15 cm in length, 150 μm i.d., Kinetex C18 core–shell particles – Phenomenex, Inc.). The eluted peptides were detected using an LTQ-Orbitrap hybrid mass spectrometer. MS/MS fragmentation was performed using collision-induced dissociation (CID) with an activation Q of 0.250, an activation time of 30.0 ms, and an isolation width of 1.0 Da. The searches were performed against the M. tuberculosis proteome including the sequences of MtCDA containing the substitutions in position 47 (E47D, E47L, E47Q). We allowed a tolerance of 10 ppm, a fragment tolerance of 0.8 Da, static carbamidomethylation on cysteines, and dynamic oxidation on methionine residues.Native polyacrylamide gel electrophoresis
Native-PAGE (N-PAGE) was carried out as described by Schägger and Gallagher.34,35 Acrylamide 4–15% linear gradient was used for gel electrophoresis. Prior to electrophoresis, proteins at 0.7 mg mL−1 concentrations were prepared in a non-denaturing sample buffer (0.5 M Tris–HCl, pH 6.8, glycerol 87%, 1 mg bromophenol blue), samples were not heated, and run in Mini-PROTEAN® 3 system (BioRad), by applying a constant milliamps at 18 mA for 3 h at 4 °C. Apparent molecular mass of bands was determined by using standard proteins with the following proteins with known molecular masses: bovine serum albumin (BSA) (monomeric form 66 kDa and dimeric form 132 kDa) and ovalbumin (monomeric form 45 kDa), these standards proteins were prepared in the same manner as the sample. Gel was stained with Coomassie Brilliant Blue R-250.Zinc analysis
Samples of recombinant mutants and wild-type MtCDA (∼2 mg mL−1) were analyzed for zinc content by inductively coupled plasma optical emission spectroscopy (ICP-OES); using a ICP-OES Perkin Elmer Optima 4300 DV (Perkin Elmer Sciex, Canada).Enzyme assays
The E47D mutant activity assay was performed as previously described for the wild-type MtCDA.19 In short, the time-dependent decrease in absorbance at 282 nm upon conversion of cytidine (εcytidine = 3.6 M−1 cm−1) into uridine or deoxyuridine was continuously monitored by a UV-2550 UV/visible spectrophotometer (Shimadzu) at 25 °C. The E47D enzyme velocity was evaluated for cytidine concentrations ranging from 100 to 800 μM.Reverse-phase HPLC had to be used to monitor the slow conversion of cytidine to uridine by the E47Q mutant enzyme. Mixtures containing Tris–HCl 50 mM, pH 7.5, mutant enzyme (0.24 mg mL−1), and cytidine (50 μM to 1800 μM) were incubated at 25 °C. At six timed intervals, aliquots (200 μL) were boiled for 3 min to stop reaction and centrifuged at 10600× g for 3 min. The supernatant (100 μL) was injected onto a reverse-phase C-18 HPLC column (0.46 × 25 cm, Amersham Bioscience) and eluted with water (0.5 mL min−1). Cytidine and uridine were separated with good resolution, having retention times of 14.7 and 27.6 min, respectively. Elution of substrate and product was monitored at 260 nm, and the integrated peak area of the product was compared with standard solutions of both cytidine and uridine.36
One enzyme unit is defined as the amount of enzyme which catalysis the deamination of 1 μmol of cytidine per minute at 25 °C.
The initial velocity method was used to calculate the apparent steady-state kinetic parameters. The initial velocity (v0) of each reaction was calculated by the linear regression of substrate concentration versus time. Hyperbolic saturation curves were fitted by nonlinear regression analysis to the Michaelis–Menten equation (eqn (1)), in which v0 is the initial velocity, Vmax is the maximal rate, [S] is the substrate concentration, and KM is the Michaelis–Menten constant. The values for the apparent kinetic constants were determined by non-linear regression using Sigma Plot version 11.0 software (Systat Software, Inc., San Jose California USA).
 | (1) |
The value for the catalytic rate constant (kcat) was determined from eqn (2), in which [E]0 represents the total concentration of enzyme subunits (as there is one binding site per subunit).
 | (2) |
pH profile
All the buffers used to determine the kinetic constants contain 100 mM 2-(N-morpholino)-ethanesulfonic acid (MES)/Hepes/2-(N-cyclohexylamino)-ethanesulfonic acid (CHES) buffer mixture.37 The pH vs. activity profiles of the E47D mutant enzyme was determined over the range of pH 4–11 using a cytidine substrate. The procedures for obtaining kinetic parameters were the same as those for the steady-state kinetic analysis as described above. Profiles were generated by plotting the either the logkcat or logkcat/KM versus pH plot.
Crystal structure determination
Crystals of MtCDA mutants E47Q and E47D were grown in hanging drops at 18 °C, using a protein solution at 6 mg mL−1 (E47Q) and 10 mg mL−1 (E47D) in 20 mM Tris HCl pH 7.5. Drops contained 1 μL of protein and 1 μL of the reservoir (0.1 M HEPES pH 7.5 and 4.3 M sodium chloride) solutions. Data collections were carried out at 100 K in a stream of liquid nitrogen gas (Oxford Cryo Systems). Crystals formed in the C2221 space group with cell dimensions of about: a = 66 Å, b = 77 Å, c = 111 Å (Table S1†). Data to 1.8 Å were collected using synchrotron radiation with λ = 1.459 Å at the MX2-beamline in LNLS/Campinas/Brazil. X-ray intensities and data reduction were evaluated using iMosflm/Scala of the CCP4 suite.38 Molecular replacement was carried out using Phaser and the wild type MtCDA structure as starting model (PDB accession code 3IJF).19 Structural modeling and real space refinement were carried out using Coot model-building tools for molecular graphics,39 and refinement of macromolecular structures using Refmac5 by the maximum-likelihood method.40 Atomic coordinates and structure factors were deposited at the Protein Data Bank (PDB codes: 4WIF and 4WIG for, respectively, the E47Q and E47D MtCDA mutants).Acknowledgements
This work was supported by funds awarded by Decit/SCTIE/MS-MCT-CNPq-FNDCT-CAPES to National Institute of Science and Technology on Tuberculosis (INCT-TB) to D.S.S. L.A.B. and D.S.S. also acknowledge financial support awarded by FAPERGS-CNPq-PRONEX-2009. L.A.B. (CNPq, 520182/99-5), D.S.S. (CNPq, 304051/1975-06), are Research Career Awardees of the National Research Council of Brazil (CNPq).V.R.J acknowledges a scholarship awarded by FAPERGS-CAPES (DOCFIX, 05/2013). Z.A.S.Q. acknowledges a scholarship awarded by CAPES.References
- World Health Organization, Global tuberculosis report 2013, WHO Press, Geneva, 2013 Search PubMed.
- E. L. Corbett, C. J. Watt, N. Walker, D. Maher, B. G. Williams, M. C. Raviglione and C. Dye, Arch. Intern. Med., 2003, 163, 1009–1021 CrossRef PubMed.
- J. E. Gomez and J. D. McKinney, Tuberculosis, 2004, 89, 29–44 CrossRef PubMed.
- G. A. O'Donovan and J. Neuhard, Bacteriol. Rev., 1970, 34, 278–343 Search PubMed.
- B. A. Moffatta and H. Ashiharab, The Arabidopsis Book, 2002, vol. 1, p. e0018 Search PubMed.
- J. Starck, G. Källenius, B. Marklund, D. I. Andersson and T. Akerlund, Microbiology, 2004, 150, 3821–3829 CrossRef CAS PubMed.
- C. W. Carter, Biochimie, 1995, 77, 92–98 CrossRef CAS.
- S. J. Chung, C. Fomme and G. Verdine, J. Med. Chem., 2005, 48, 658–660 CrossRef CAS PubMed.
- L. Betts, S. Xiang, S. A. Short, R. Wolfenden and C. W. Carter, J. Mol. Biol., 1994, 235, 635–656 CrossRef CAS PubMed.
- M. J. Snider, S. Guanitz, C. Ridway, S. A. Short and R. Wolfenden, Biochemistry, 2000, 39, 9746–9753 CrossRef CAS PubMed.
- D. C. Carlow, A. A. Smith, C. C. Yang and S. A. Short, Biochemistry, 1995, 34, 4220–4224 CrossRef CAS.
- M. J. Snider, L. Reinhardt, R. Wolfenden and W. W. Cleland, Biochemistry, 2002, 41, 415–421 CrossRef CAS PubMed.
- C. S. Harold, RNA and DNA editing: Molecular mechanisms and their integration into biological systems, 2008, vol. 1, pp. 232–238 Search PubMed.
- B. Weiss, J. Bacteriol., 2007, 189, 7922–7926 CrossRef CAS PubMed.
- D. C. Carlow, Biochemistry, 1999, 38, 12258–12265 CrossRef CAS PubMed.
- S. E. Faivre-Nitschke, J. M. Grienenberger and J. M. Gualberto, Eur. J. Biochem., 1999, 3, 896–903 CrossRef.
- S. Vincenzetti, A. Cambi, J. Neuhard, E. Garanttini and A. Vita, Protein Expression Purif., 1996, 8, 247–253 CrossRef CAS PubMed.
- E. Johansson, N. Mehlhede, J. Neuhard and S. Larsen, Biochemistry, 2002, 41, 2563–2570 CrossRef CAS PubMed.
- Z. A. Sánchez-Quitian, C. Z. Schneider, R. G. Ducati, W. F. de Azevedo Jr, C. Bloch, L. A. Basso and D. S. Santos, J. Struct. Biol., 2010, 169, 413–423 CrossRef PubMed.
- Z. A. Sánchez-Quitian, L. F. Timmers, R. A. Caceres, J. G. Rehm, C. E. Thompson, L. A. Basso, W. F. de Azevedo Jr and D. S. Santos, Arch. Biochem. Biophys., 2011, 509, 108–115 CrossRef PubMed.
- S. Vicenzetti, B. Quadrini, P. Mariani, G. D. Sanctis, N. Cammertoni, V. Polzonetti, S. Puciarelli, P. Natalini and A. Vita, Proteins, 2008, 70, 144–156 CrossRef PubMed.
- H. P. Sørensen and K. K. Mortensen, J. Biotechnol., 2005, 115, 113–128 CrossRef PubMed.
- B. Magalhães, C. P. Pereira, L. A. Basso and D. S. Santos, Protein Expression Purif., 2002, 26, 59–64 CrossRef.
- R. A. Copeland, Enzymes: A Practical Introduction to Structure, Mechanism, and Data Analysis, Wiley-VCH, New York, 2000, ch. 5, pp. 120–123 Search PubMed.
- D. C. Carlow, S. A. Short and R. Wolfenden, Biochemistry, 1998, 37, 1199–1203 CrossRef CAS PubMed.
- G. L. Holliday, J. B. O. Mitchell and J. M. Thornton, J. Mol. Biol., 2009, 390, 560–577 CrossRef CAS PubMed.
- G. J. Bartlett, C. T. Porter, N. Borkakoti and J. M. Thornton, J. Mol. Biol., 2002, 324, 105–121 CrossRef CAS.
- S. N. Ho, H. D. Hunt, R. M. Horton, J. K. Pullen and L. R. Pease, Gene, 1989, 77, 51–59 CrossRef CAS.
- U. K. Laemmli, Nature, 1970, 227, 680–685 CrossRef CAS.
- M. M. Bradford, Anal. Biochem., 1976, 72, 248–254 CrossRef CAS.
- G. R. Grimsley and C. N. Pace, Current Protocols in Protein Science, 2003, 3.1.1–3.1.9 Search PubMed.
- A. A. Klammer and M. J. MacCoss, J. Proteome Res., 2006, 5, 695–700 CrossRef CAS PubMed.
- R. L. Moritz, CSH Protocols, 2007 DOI:10.1101/pdb.prot4578.
- H. Schägger and G. Von Jagow, Anal. Biochem., 1987, 166, 368–379 CrossRef.
- S. R. Gallagher, Current Protocols in Cell Biology, 2001, 5, 6.5.1–6.5.11 Search PubMed.
- M. G. Williams, J. Palandra and E. M. Shobe, Biomed. Chromatogr., 2003, 17, 215–218 CrossRef CAS PubMed.
- P. F. Cook and W. W. Cleland, in Enzyme Kinetics and Mechanism, Garland Science Publishing, New York, 2007, ch. 10, pp. 325–366 Search PubMed.
- Collaborative Computational Project, Number 4, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1994, 50, 760–763 CrossRef PubMed.
- P. Emsley and K. Cowtan, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2004, 60, 2126–2132 CrossRef PubMed.
- G. N. Murshudov, A. A. Vagin and E. J. Dodson, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1997, 53, 240–255 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra13748e |
| This journal is © The Royal Society of Chemistry 2015 |