
QSAR-guided semi-synthesis and in vitro validation of antiplasmodial activity in ursolic acid derivatives†
Abstract
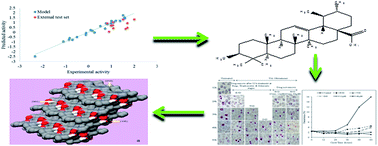
As a part of our antimalarial drug discovery programme, a quantitative structure–activity relationship (QSAR) model was developed for the prediction of antiplasmodial activity in ursolic acid (UA) derivatives, followed by the wet laboratory semi-synthesis of virtually active derivatives and their in vitro biological evaluation. The QSAR model was developed by a forward stepwise multiple linear regression method using a leave-one-out approach, which showed a 96% prediction accuracy. The most active virtual derivatives of UA were semi-synthesized and their in vitro evaluation showed a similar antiplasmodial activity to the predicted one, thus validating the QSAR model. Further, the in silico mode of action was studied on β-hematin and subsequently confirmed by in vitro FPIX biomineralization inhibition activity. Finally, the in silico ADME/T properties and cytotoxicity against Vero cells using an MTT assay were studied. Out of eight derivatives, the two UA-18 and UA-21 showed significant dose-dependent antiplasmodial activity in arresting the progress of P. falciparum erythrocytic cycle. Furthermore, the in silico and in vitro mode of action studies of these derivatives showed comparable binding energies and percentage inhibitions on β-hematin to that of the standard drug chloroquine (CQ). The in silico ADME/T analysis of UA and its derivatives did not show CYP2D6 inhibition, hepatotoxicity nor mutagenicity, but showed a poor solubility and poor human intestinal absorption. These findings may be of immense importance in antiplasmodial drug development from an inexpensive and widely available natural product, i.e. ursolic acid.
 Please wait while we load your content...
Please wait while we load your content...

