
Novel integrated carbon particle based three dimensional anodes for the electrochemical degradation of reactive dyes†
Abstract
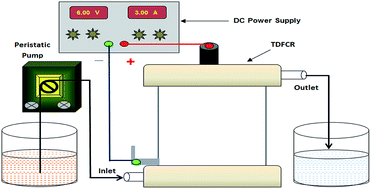
Three-dimensional carbon bed electrochemical reactors have been recently applied for the degradation of several organic pollutants. However, the carbon particles in such reactors slowly undergo attrition. We fabricated a novel flow-through three-dimensional anode using granular activated carbon (GAC) particles and polyvinylidene fluoride (PVDF) binder that potentially avoids such attrition. Optimization of the composition of GAC and PVDF with respect to mechanical integrity and electrical conductivity is reported. The anodes were tested in the electro oxidation of the reactive dyes: Reactive Orange-16 (RO-16), Reactive Red-2 (RR-2), and Reactive Blue-4 (RB-4). A tentative mechanism of dye degradation was proposed based on the observed role of the supporting electrolyte and the cyclic voltammetric, UV-vis, FT-IR and GC-MS data. The decolorization efficiencies were 75 ± 3, 81 ± 5 and 88 ± 4% for RB-4, RO-16 and RR-2, respectively. The integrated 3-D anodes are advantageous because of the absence of carbon attrition, which is otherwise found when a bed of GAC is used in the electrochemical reactors.
 Please wait while we load your content...
Please wait while we load your content...

