
Screening of β-hairpin peptide-engrafted 1,2,3-triazoles to identify APEH enzyme inhibitors†
A. Sandomenico‡
*ab,
V. Celentano‡a,
L. D. D'Andreaab,
G. Palmieric and
M. Ruvo*ab
aInstitute of Biostructure and Bioimaging, National Research Council (CNR-IBB), Via Mezzocannone 16, 80134 Naples, Italy. E-mail: annamaria.sandomenico@gmail.com; menotti.ruvo@unina.it; Fax: +39-081-2534574; Tel: +39-081-2536644
bCIRPEB-University of Naples Federico II, Naples, Italy
cInstitute of Biosciensces and BioResources, National Research Council (CNR-IBBR), Naples, Italy
First published on 6th January 2015
Abstract
APEH catalyses the removal of N-terminal acetylated amino acids from proteins destined to be degraded and is now recognized as a new therapeutic target for several diseases. New APEH inhibitors having triazole-based structures have been recently reported. On this basis we have screened a set of click-generated cyclic peptides, previously investigated for peptide conformational stability studies, as possible novel enzyme inhibitors. We have found a clicked peptide, NHB3.3, that inhibits APEH activity and structure–activity studies highlighted that APEH inhibition is mediated by the spatial organization of the triazole ring and by its orientation and distance from the peptide scaffold, whose structural integrity, in turn, also plays a relevant role. In conclusion, our findings confirm that 1,2,3 triazoles are privileged pharmacophores for specific serine protease inhibitors and provide structural insights exploitable for modulating their inhibition activity.
Introduction
APEH (EC 3.4.19.1), namely acyl-amino-acid-releasing enzyme (or acylpeptide hydrolase or acyl-amino-acyl peptide hydrolase), is a relatively new member of the prolyl oligopeptidase (POP) family of serine peptidases (clan SC, family S9).1 As most of enzymes belonging to the POP family, APEH hydrolyses short peptide substrates and presents a canonical α/β hydrolase fold, together with the Ser–Asp–His catalytic triad covered by an unusual β-propeller.2 So far, this class of enzymes has been mainly found in a number of eukaryal and archaea organisms as well as in a psychrotollerant bacterium.3,4 APEH catalyses the removal of acetyl-amino acids from the N-terminus of short peptides and cytoplasmic proteins. For these properties the enzyme is attracting a vast attention for the potential role it can play in the complex mechanisms regulating protein turnover. Although the physiological role of APEH in cells is still unclear, several experimental evidences have highlighted the impact the activity of this enzyme may have on the fate of cancer cells.Several reports have indeed suggested its implication in the molecular mechanisms of protein degradation5,6 and its function as secondary antioxidant defence system against proteins damaged by oxidative stress7 and as modulator of cancer progression.8–12
The identification of potent and selective APEH inhibitors, for these reasons, is therefore become an important focus, because they serve as tools to investigate the enzyme downstream effects and may be employed in those therapeutic applications requiring a down-regulation of APEH activity.7 Previously, we identified APEH inhibitors by screening a set of synthetic peptides designed on the reactive-site loop (RSL) of an APEH protein inhibitor.6 A competitive inhibitor (Ki = 1.00 ± 0.02 μM) was derived from the first endogenous inhibitor SSCEi (Sulfolobus solfataricus Chymotrypsin–Elastase inhibitor), and was employed to investigate the APEH role in the protein degradation machinery and its correlation with the UPS (ubiquitin–proteasome-system). By screening a random peptide library13 we also selected a modified tetrapeptide, named CF3-lmph, showing an unusual uncompetitive inhibition activity. CF3-lmph was able to block the APEH activity in cells without significant effect on cell proliferation and caspase activity.13 The different effects produced by the two APEH inhibitors on cells proliferation, suggested a possible intriguing relationship between the mechanism of APEH inhibition and proteasome activity. Recently, 1,2,3-triazole urea-derivatives have also been identified as potent inhibitors of APEH and other serine hydrolases.14 On this basis, we have reasoned that click-generated peptide-engrafted triazoles could inhibit the enzyme and that peptide sequence and structure might modulate such activity. To test this hypothesis we screened a set of 1,4-disubstituted 1,2,3-triazole engrafted on a modified Trpzip2 peptide scaffold previously generated for peptide conformational stability studies.15 These compounds consisted of a triazole ring attached to a peptide chain through the spacers provided by the side chains of different precursors. For this purpose, we used azide- and propargyl-bearing residues in which the number and position of methylene units were varied (Fig. S1†). After screening the first set of molecules, we have prepared and tested new analogues to dissect the relative contribution of the different molecular substructures to APEH recognition and inhibition. The best inhibitor has then been characterized in terms of specificity and mechanism of inhibition toward APEH.
Results and discussion
Library preparation and characterization
All crude peptidomimetics were obtained with average yields of 50–70% and after RP-HPLC purification homogeneous products were isolated. Experimental molecular masses were all consistent with the expected values (Table S1†). Unprotected linear peptides were cyclized (clicked) via a CuAAC reaction as described in the Methods; since the triazole ring formation does not involve a change of the molecular weight, cyclization was monitored via RP-HPLC by the formation of products with significantly shorter retention times compared to the corresponding linear products and with the same molecular masses15 (Table S1†). Conversions were all quantitative. The NHB3.3_open peptide was obtained by treatment with trypsin of the NHB3.3 cyclic precursor. This treatment afforded only one HPLC peak with a mass corresponding to the open hydrolyzed peptide. The cleaved peptide was purified to homogeneity by RP-HPLC.Screening of clicked peptidomimetics
 | ||
| Fig. 1 Top: generic molecular structure of the 1st generation library (entries 1–15) of NHB clicked peptidomimetics. Bottom: nomenclature used to identify the tested compounds; x and y indicate the number of methylene groups on the 1,4-disubstituted 1,2,3-derived cyclic molecules. Detailed molecular structures of all compounds are reported as ESI.† | ||
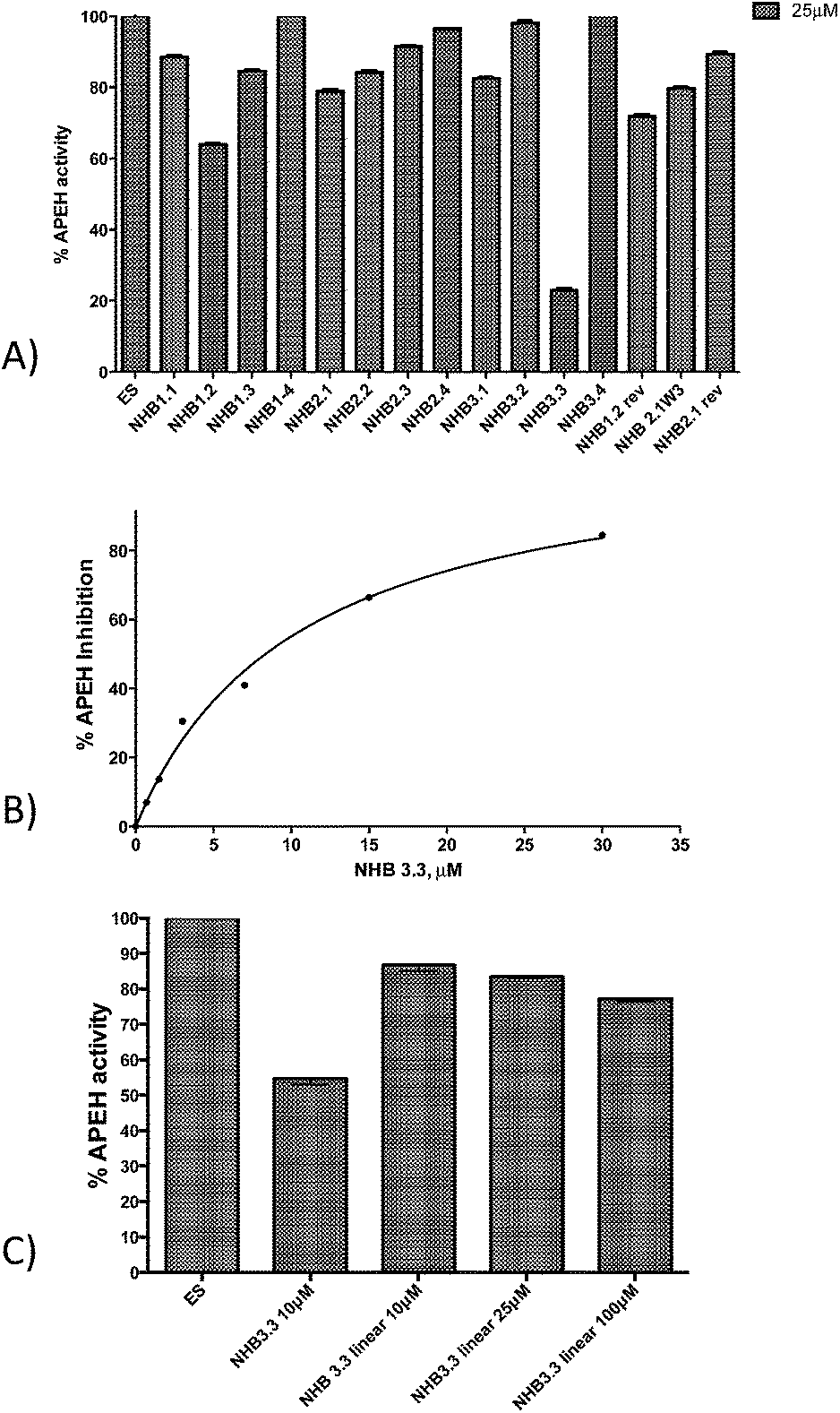 | ||
| Fig. 2 (A) Inhibition of APEH activity by clicked peptidomimetics included the 1st generation library of NHB compounds. Peptides were tested at the concentration of 25 μM. (B) Dose–response assay for the most active compound. The IC50 value (10.5 ± 1.7 × 10−6 M) was determined by fitting the experimental data with the GraphPad Prism software, using a nonlinear curve-fitting method and a simple binding isotherm equation {%I = %Imax[I]/(IC50 + [I])}. (C) Inhibition of APEH activity by clicked NHB3.3 and its corresponding unclicked (linear) precursor tested at the concentration of 10, 25 and 100 μM. | ||
Two additional compounds, NHB1.2 and NHB1.2rev, exhibited a significant inhibition, but they reduced APEH activity for only 40% and 30%, respectively. Next, a dose–response assay was carried out using NHB3.3 at increasing concentrations between 0 and 30 μM. As reported in Fig. 2B, APEH inhibition followed a hyperbolic trend, reaching 80% at the highest inhibitor concentration. The extrapolated IC50 was 10.5 ± 1.7 × 10−6 M. Remarkably, the NHB3.3 unclicked linear variant, lacking of the triazole ring, when tested at 10 μM, 25 μM and 100 μM, exhibited only a poor inhibition (13%, 16% and 22%, respectively; Fig. 2C) confirming the critical role exerted by the 1,2,3-triazole moiety in blocking APEH activity.14 In addition, the very poor activity displayed by other library components bearing the triazole ring, demonstrated that inhibition of APEH by NHB3.3 also depends on specific structural properties beyond the presence of the heterocycle. Data thus suggest that the specific peptide structure, the triazole moiety and, more importantly, the spatial orientation induced by the length of 1,4 substituents, together contribute to the inhibiting activity toward APEH.
 | ||
| Fig. 3 Inhibition of APEH activity by the set of 2nd generation of NHB3.3 peptidomimetics and the corresponding unclicked linear variants tested at 10 μM. | ||
 | ||
| Fig. 4 Dose–response assay for NHB3.3_C (A) NHB3.3_N (B) and NHB3.3_CN (C) carried out at concentrations between 5 and 150 μM. | ||
Mechanism of inhibition
To further investigate the role played by the peptide structure in APEH inhibition, we generated and tested the acyclic variants NHB3.3_Δturn and NHB 3.3_open. We found that both the absence of the β-turn sequence and splitting of the molecule on the internal lysine, negatively affected the peptide activity, which, at the maximum concentration of 100 μM, was 21% and 31% (Fig. 5A), respectively. NHB3.3_open was also dose-dependently tested at concentrations between 0 and 200 μM, confirming that less than 30% inhibition was afforded by this compound, even at very high concentrations (Fig. 5B). To assess the role of the N-terminal free amino group, we finally tested the activity of the acetylated NHB3.3 variant (NHB3.3Ac). Strikingly, capping the free amino group completely ablated the inhibition properties, suggesting a direct involvement of this group in enzyme recognition or a strong influence of the peptide net charge (Fig. 6). | ||
| Fig. 5 (A) Inhibition of APEH activity by NHB3.3_Δturn and NHB3.3_open at 10 μM and 100 μM. (B) Dose–response assays for NHB3.3_open carried out at concentrations between 5–200 μM. | ||
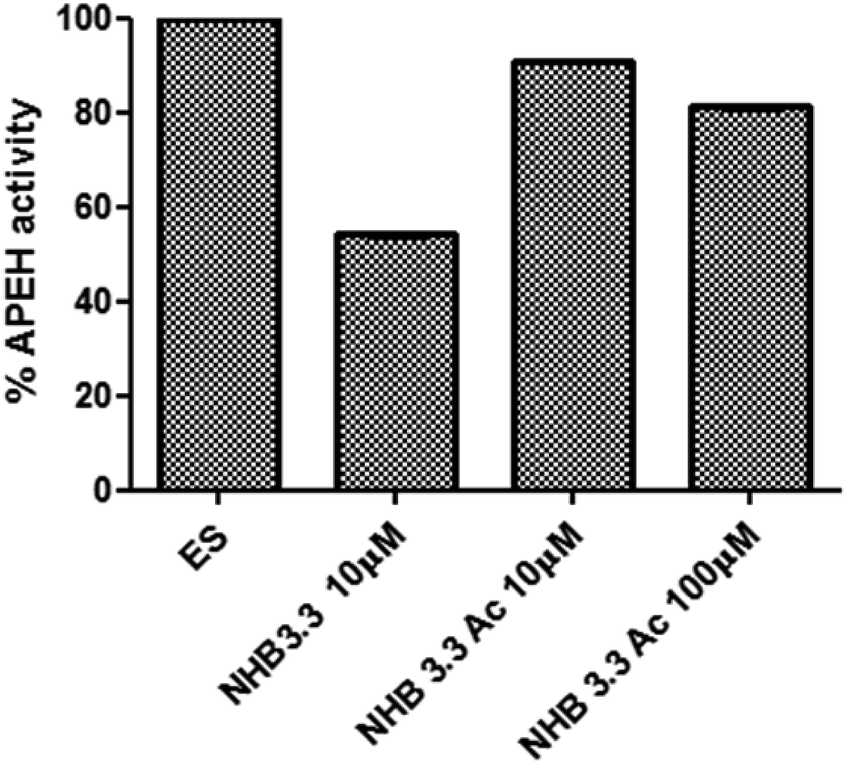 | ||
| Fig. 6 Inhibition of APEH activity by acetylated NHB3.3 variants tested at the concentrations of 10 μM and 100 μM. | ||
To further gain insights into the recognition, we explored the inhibition mechanism of NHB3.3 toward APEH purified from porcine liver. The enzyme was pre-incubated with the inhibitor at two concentrations and the residual enzymatic activity was assessed in the presence of increasing concentrations of the AANA substrate. The Lineweaver–Burk plots where the y-intercept is equivalent to the inverse of Vmax and the x-intercept represents −1/Km, reported in Fig. 7A, shows that the lines obtained at different inhibitor concentrations, do not intersect on the 1/Vmax axis, suggesting that the compound acts as a non-competitive inhibitor. The inhibition constants Ki was estimated by the Lineweaver–Burk equation (Fig. 7B), which provided a value of 10.8 ± 3.7 × 10−6 M, in close agreement with the IC50 value obtained in the dose–response inhibition assay. Also a Km of 100 μM could be determined by this approach.
 | ||
| Fig. 7 (A) Inhibition kinetic analysis. APEH was incubated without (ES) or with the NHB3.3 at 15 μM (EI 15 μM) and 30 μM (EI 30 μM)) and assayed at increasing concentrations of substrate (5–500 μM). The reciprocals of the rate of the substrate hydrolysis for each inhibitor concentration were plotted against the reciprocals of the substrate concentrations. (B) By a plot of (1 + I/Ki) against the NHB3.3 concentration a dissociation constant (Ki) value was extrapolated which is consistent with the IC50 determined previously. | ||
Inhibition specificity
We next determined the NHB3.3 specificity toward APEH. The activity of the peptide inhibitor was thus probed against a panel of serine proteases, including elastase, trypsin, chymotrypsin and subtilisin. The chymotrypsin-like activity of proteasome was also probed using a suitable chromogenic substrate. Results are summarized in Table 1.| 100 μM | 50 μM | 25 μM | |
|---|---|---|---|
| Trypsin | No inhibition | No inhibition | No inhibition |
| Elastase | No inhibition | No inhibition | No inhibition |
| Subtilisin | No inhibition | No inhibition | No inhibition |
| Chymotrypsin | 60% | 30% | 15% |
| Proteasome | 63% | 45% | 30% |
| APEH | 100% | 100% | 80% |
The triazole-peptide was inactive toward trypsin, elastase and subtilisin, whereas it inhibited chymotrypsin by 30% and proteasome by 45% at 50 μM, a concentration sufficient to completely abolish APEH activity (Table 1). Data are thus suggestive of a strong ability of the inhibitor to discriminate between APEH and other proteases showing similar mechanisms, though it is capable to partially interfere with the chymotrypsin activity shared by chymotrypsin itself and one of the proteasome subunits (β5).
Discussion
In this study, we have screened a set of click-generated triazole-containing cyclic peptidomimetic to select inhibitors of the enzyme APEH, which is now recognized as a new therapeutic target for several diseases, including cardiovascular diseases, cancer, inflammation, haematological diseases, neurological and urological diseases.3 All the clicked compounds have a common Trpzip2-like peptide structure, whereas the linkers connecting the peptide to the triazole ring varied in length affecting the β-hairpin conformational stability.15 We selected the NHB3.3 clicked peptide as the best APEH inhibitor among all the tested analogues. NHB3.3 bears on both β-hairpin strands three methylene groups deriving from the triazole precursors bis-homopropargylglycine (Bpg) and l-δ-azido-ornitine (Orn(N3)). NHB3.3 dose-dependently blunts APEH activity with an 80% maximum inhibition reached at 30 μM and with an IC50 of 10.5 ± 1.7 μM. Remarkably, enzyme inhibition occurs through a non-competitive mechanism, therefore enzyme recognition likely takes place on a site not overlapped with the catalytic pocket and could involve an allosteric mechanism. Our findings are in agreement with previous data by Adibekian et al.14 showing that APEH is inhibited by substituted 1,2,3-triazoles. Compared to N-heterocyclic ureas triazole-based inhibitors, which have subnanomolar activities,14 our inhibitor shows a much lower inhibition efficiency. It should be underscored that irreversible hydrolase inhibitors do interact covalently with the target enzyme, showing tremendously high potencies. However, while potency is undoubtedly the most important aspect, it should be considered that they are not generally well accepted for eventual clinical use, especially when the target biology is still controversial and not well understood as in the case of APEH. We therefore believe that reversible inhibitors, as those potentially obtainable from those we report here, would be invaluable tools to further investigate the therapeutic potential of APEH.A step forward in the process of improving our inhibitors' potency will be the introduction of a second recognition center, which would ideally synergize with the triazole primary unit to interact with and reversibly block the enzyme.14,16 The peptide structure of NHB3.3 may serve for this purpose, acting as a support for introducing such auxiliary anchoring point.
In this instance it should be underscored that the peptide structure and the triazole moiety together contribute to enzyme recognition and inhibition, therefore the site for modification must be carefully chosen. Our structure–activity relationship study on NHB3.3 has indeed confirmed that the intact triazole ring and a specific spatial orientation are both required for activity. The peptide structure clearly functions as a scaffold for properly positioning the ring, therefore the secondary structure has a primarily importance for this purpose.
As reported by Celentano et al.,15 NHB3.3 adopts a β-hairpin structure, but among those studied at structural level, it is not the one with the highest β-hairpin content. On the other hand, the peptide NHB2.1, which showed the highest β-hairpin content,15 only moderately (around 20%) affects APEH activity. This suggests that the conformational stability of the peptide β-hairpin structure is not a requirement for activity, rather a partial flexibility seems to favour enzyme recognition. Indeed, increasing the linker length (NHB3.4 vs. NHB3.3) the peptide inhibitory activity is completely abolished, suggesting that the size of the bridge supporting the triazole ring could be critical. It has to be noted that peptides with four methylene groups are always the worst inhibitor for each NHB series with a fixed x number (i.e. NHB1.1, NHB1.2, NHB1.3, NHB1.4).
Fixed the optimal orientation between the triazole ring and the peptide chain (such as in NHB3.3), it seems that the structural integrity of the β-hairpin also plays a role in APEH inhibition. Removal of the turn residues (NHB3.3_Δturn), turn opening (NHB3.3_open), deletion of peptide termini (NHB 3.3_N, NHB3.3_C or NHB3.3_NC) renders NHB3.3 ineffective or reduces APEH inhibition. It is therefore reasonable to hypothesize that these backbone modifications abolish the inhibition activity, by drastically modifying the β-hairpin secondary structure.
Taken together these experimental observations confirm that the inhibitory effect of peptide NHB3.3 depends on the spatial organization of the triazole ring, its orientation and distance with respect to the peptide chain and on the structural integrity of the peptide scaffold.
Conclusions
In conclusion, our findings confirm that 1,2,3-triazoles are key components of a pharmacophoric structure common to serine protease and more specifically APEH inhibitors and that such structural module is exploitable to finely regulate the inhibition activity. Given the increasing importance of APEH in several diseases and its connection with the molecular mechanisms of protein turnover, peptide-linked 1,2,3-triazole derivatives can represent versatile tools for developing more potent and specific APEH-inhibitors.Experimental
Materials
Protected amino acids for the synthesis of peptides and the activators N-hydroxybenzotriazole (HOBT) and O-benzotriazole-N,N,N′,N′-tetramethyl-uronium-hexafluoro-phosphate (HBTU) coupling agents were from Merck except Fmoc-l-Dap(N3)–OH, Fmoc-l-Dab(N3)–OH, Fmoc-L-Orn(N3)–OH which were from Iris Biotech GmbH. Solvents for peptide synthesis and purification, acetic anhydride, ascorbic acid and copper(II) sulfate pentahydrate were from Sigma-Aldrich (Milan, Italy). Piperidine was purchased from Biosolve; N,N-diisopropylethylamine (DIPEA) was provided by Romil. Porcine liver APEH was obtained from Takara BIO INC. (Shiga Japan). Acetyl-Ala-pNA was from Bachem (Laufelfingen, Switzerland). Elastase, subtilisin, trypsin, and chymotrypsin were from Sigma-Aldrich (Milano, Italy). Purified human erythrocyte proteasome was from Boston Biochem (Boston, USA). N-Succinyl-Ala–Ala–Pro–Phe p-nitroanilide (Suc-AAPF-pNA). Nα-Benzoyl-L-arginine 4-nitroanilide hydrochloride (l-BAPA) and N-succinyl-Leu–Leu–Val–Tyr-7-amido-4-methylcoumarin substrate were from Sigma-Aldrich (Milano, Italy).Library preparation and characterization
Given the occurrence of 1,2,3-triazole moieties in small molecule inhibitors of APEH,14 we have initially used a series of available short peptides side chain cyclized via Cu(I)-catalyzed azide alkyne cycloaddition (CuAAC) reactions. After the click reaction, compounds incorporated a 1,4-disubstituted 1,2,3-triazole-ring engrafted on a Trpzip2-derived peptide scaffold15 in which two residues in non-hydrogen bonded (NHB) positions, Trp4 and Trp9, were replaced with a homologue series of alkyne and azide amino acids. The basic structures of the click peptides used in this study and the molecular formulas of the unnatural amino acids used are reported in Fig. S1–S3 supplied as ESI.†Unnatural amino acids with different side chain lengths were introduced on the Xaa and Yaa positions. Specifically, on position Xaa we introduced alkyne propargylglycine (Pra), homopropargylglycine (Hpg), bishomopropargylglycine (Bpg), while in position Yaa we placed L-β-azidoalanine (Dap (N3)), L-γ-azido-homoalanine (Dab (N3)), l-δ-azido-ornitine (Orn (N3)) and N-ε-azido-L-lysine (Lys (N3)). The different combinations of Xaa and Yaa are reported in Fig. 1 and S1.† The first group of resulting 12 click compounds were named 1st generation library of NHB-clicked peptides. To further investigate the inhibition properties of this class of compounds, we also screened in the same set three additional peptides, named NHB1.2rev, NHB2.1rev and NHB-W3.15 In NHB1.2rev and NHB2.1rev, the positions of alkyne- and azide-amino acids were exchanged compared to the NHB1.2 and NHB2.1 parent peptides (Fig. 1, S2 and S3†), peptides from number 1 to 15, while in NHB-W3 the tryptophan originally on position 2, was placed on position 3. To investigate the influence of selected residues from the Trpzip2-like peptide sequence on APEH recognition and inhibition, after the screening of the first generation library, we designed and synthesized a second set of NHB3.3-derived compounds (2nd generation of NHB-clicked peptides (Fig. 1 and S3†) peptides from number 16 to 20). In particular, we generated the unclicked linear variant (NHB3.3_linear), and three chain shortened peptide variants where the C-terminal threonine, valine and lysine (NHB3.3_C), the N-terminal serine and threonine (3.3_N) or both (NHB3.3_NC) were ablated. Tryptophan was maintained in all compounds to perform UV quantification. We also designed and synthesized a non-cyclic NHB3.3 variant without the internal β-turn sequence Glu–Asn–Gly–Lys (NHB3.3_Δturn). An additional ring open variant of clicked NHB3.3 was obtained by splitting the bond between lysine and Orn(N3) with trypsin (NHB3.3_open).
All peptides were synthesized on solid phase by standard Fmoc chemistry using a Rink Amide MBHA resin (0.54 mmol g−1 substitution) as solid support, as it releases peptides amidated at C-terminus upon acid treatment. All couplings were performed twice for 20 min by using an excess (2.5 equivalent) of each amino acid derivative. Amino acids were activated in situ by 2.49 equivalents of HOBt/HBTU and 5 equivalents of DIPEA. Fmoc deprotection was performed with 30% piperidine in DMF (5 min, twice). After the last coupling, unreacted N-terminal amino groups were acetylated with a solution of 2.0 M acetic anhydride, 0.06 M HOBt, 0.55 M DIPEA in NMP (5 min). Peptide cleavage from the solid support and simultaneous removal of all protecting groups from amino acid side chains was carried out by suspending the fully protected compound-resins in a TFA/H2O/TIS (95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.5:2.5) mixture for 3 h. Crude products were precipitated by addition of cold diethyl ether after filtration of the resin, washed three times with diethyl ether, collected by centrifugation, dissolved in a water/acetonitrile mixture and lyophilized. The purified linear peptides were subjected to cyclization by Cu(I)-catalyzed azide alkyne cycloaddition (CuAAC) reaction (0.5 mM peptide, 7 mM CuSO4, 6.5 mM ascorbic acid in H20/CH3OH (2:1 v/v) at r.t. for 60 min.17–19 Analytical RP-HPLC runs were carried out on an HP Agilent Series 1200 chromatograph using a Proteo C12 column (4 μm, 90 Å, 4.6 × 250 mm (Phenomenex, Torrance, CA) operated at a flow rate of 1.0 mL min−1. Preparative RP-HPLC was carried out on a Shimadzu 8A chromatograph coupled with an UV detector, using an AXIA column 50 × 21.2 mm, 4 μm (Phenomenex, Torrance, CA); the flow rate was 20 mL min−1. For all the RP-HPLC purifications the system solvent used was: H2O 0.1% TFA (solvent A) and CH3CN 0.1% TFA (solvent B). Typical linear gradients starting from 5% to 70% B in 20 min were applied; UV detectors were set at 210 nm and 280 nm. The purified clicked peptides were characterized by LC-MS using an LCQ DECA XP Ion Trap mass spectrometer equipped with an OPTON ESI source, operating at 4.2 kV needle voltage and 320 °C and coupled to a complete Surveyor HPLC system, comprising an MS pump, an autosampler and a photo diode array (PDA) detector. A Jupiter C18 column (5 μm, 300 A, 250 × 2 mm, Phenomenex) at a flow rate 0.2 mL min−1 was used in these analyses. Typical LC-MS gradients were from 5% to 70% solvent B (acetonitrile with added 0.05% TFA; solvent A was water with 0.05% TFA). NHB3.3_open was obtained by tryptic digestion. The reaction was carried out adding porcine trypsin at 1:10 w/w in 50 mM Tris–HCl pH 7.5 for 4 h at 37 °C. The reaction was stopped by acidification with H2O, 0.1% TFA. The product was analysed by LC-MS and purified to homogeneity as described above for the other compounds.
:2.5:2.5) mixture for 3 h. Crude products were precipitated by addition of cold diethyl ether after filtration of the resin, washed three times with diethyl ether, collected by centrifugation, dissolved in a water/acetonitrile mixture and lyophilized. The purified linear peptides were subjected to cyclization by Cu(I)-catalyzed azide alkyne cycloaddition (CuAAC) reaction (0.5 mM peptide, 7 mM CuSO4, 6.5 mM ascorbic acid in H20/CH3OH (2:1 v/v) at r.t. for 60 min.17–19 Analytical RP-HPLC runs were carried out on an HP Agilent Series 1200 chromatograph using a Proteo C12 column (4 μm, 90 Å, 4.6 × 250 mm (Phenomenex, Torrance, CA) operated at a flow rate of 1.0 mL min−1. Preparative RP-HPLC was carried out on a Shimadzu 8A chromatograph coupled with an UV detector, using an AXIA column 50 × 21.2 mm, 4 μm (Phenomenex, Torrance, CA); the flow rate was 20 mL min−1. For all the RP-HPLC purifications the system solvent used was: H2O 0.1% TFA (solvent A) and CH3CN 0.1% TFA (solvent B). Typical linear gradients starting from 5% to 70% B in 20 min were applied; UV detectors were set at 210 nm and 280 nm. The purified clicked peptides were characterized by LC-MS using an LCQ DECA XP Ion Trap mass spectrometer equipped with an OPTON ESI source, operating at 4.2 kV needle voltage and 320 °C and coupled to a complete Surveyor HPLC system, comprising an MS pump, an autosampler and a photo diode array (PDA) detector. A Jupiter C18 column (5 μm, 300 A, 250 × 2 mm, Phenomenex) at a flow rate 0.2 mL min−1 was used in these analyses. Typical LC-MS gradients were from 5% to 70% solvent B (acetonitrile with added 0.05% TFA; solvent A was water with 0.05% TFA). NHB3.3_open was obtained by tryptic digestion. The reaction was carried out adding porcine trypsin at 1:10 w/w in 50 mM Tris–HCl pH 7.5 for 4 h at 37 °C. The reaction was stopped by acidification with H2O, 0.1% TFA. The product was analysed by LC-MS and purified to homogeneity as described above for the other compounds.
Screening of clicked peptides
The screening was carried out by a spectrophotometric assay using porcine acyl peptide hydrolase (APEH) enzyme and the synthetic specific substrate N-acetyl-L-alanine p-nitroanilide (AANA). The reaction mixture (0.2 mL), containing pure APEH (0.5 nM) in 50 mM Tris–HCl buffer pH 7.5, was pre-incubated at 37 °C for 2 min. Then, 16 μM AANA was added. The release of p-nitroanilide (ε = 8800 M−1 cm−1) was measured by recording the absorbance increase at 410 nm on a Synergy multi-wavelength plate reader (Biotek), equipped with a thermostated compartment. Assays were performed in triplicates in 96-well polyethylene plates. The inhibition screening assay was carried out by pre-incubating each peptide (25 μM) with the enzyme for 10 min at 37 °C prior addition of the substrate. The absorbance at 410 nm was continuously monitored in each well and the slope of each straight line was obtained. Data were processed by averaging replicate values and calculating slopes by linear regression analysis. The inhibition extents (%) were determined by comparing the slopes obtained from inhibition assays with those resulting from control experiment (assumed 100%).Dose–response assays with NHB3.3 were performed at concentrations ranging between 0.7 and 30 μM with the purpose of confirming results at different concentrations and to determine IC50. The 2nd generation of NHB-clicked peptides, including both the Δturn and open variants, was dose-dependently screened at concentrations between 10 μM and 100 μM.
Dose-dependent assays were also conducted for NHB3.3_C, NHB3.3_N and NHB3.3_NC at concentrations between 5 μM and 150 μM. IC50 values were extrapolated by data fitting with the GraphPad Prism software by using a nonlinear curve-fitting method and a simple binding isotherm equation {%I = %Imax[I]/(IC50 + [I])}. All assays were performed in 96-well plates and each data point was determined as triplicate. Experiments were repeated at least two times. Data are reported as values ± standard deviation.
Mechanism of inhibition
To determine the mechanism of enzyme inhibition, APEH (0.5 nM) was pre-incubated without and with NHB3.3 at 15 μM and 30 μM and its activity was tested at increasing substrate concentrations (between 5 and 500 μM). For each inhibitor concentration, a Lineweaver–Burk double reciprocal plot was built by plotting the reciprocal of substrate hydrolysis rate as a function of the reciprocals of substrate concentrations. The inhibition constant Ki was determined by the Lineweaver–Burk equation {1/V = (Km/Vmax)(1 + I/Ki)1/S + 1/Vmax(1 + I/Ki)}.Inhibition specificity
To assess the specificity of NHB3.3 in inhibiting APEH, we tested its activity against a panel of other common serine proteases. For this purpose we used elastase, subtilisin, trypsin, chymotrypsin and proteasome with the suitable chromogenic substrates. For proteasome the chymotrypsin-like activity was probed. Reactions were performed in 1 mL of buffer. For elastase, subtilisin, trypsin, chymotrypsin and APEH, assays were performed using the specific chromogenic substrates reported below, all having a p-nitroanilide reporter (pNA). Enzymatic activities were measured on a Cary 100 Scan (Varian) UV/Vis spectrophotometer, equipped with a thermostated cuvette compartment by recording the absorbance increase at 410 nm due to p-nitroaniline release. The chymotrypsin-like activity of proteasome was determined by a fluorimetric assay were the reporter was amido-4-methylcoumarin. In detail, elastase and subtilisin activities were determined using a 400 nM solution of enzyme in 25 mM Tris–HCl pH 8.0 and 100 μM Suc-AAPF-pNA substrate. Trypsin was assayed using the enzyme at 160 nM in 50 mM Tris–HCl buffer pH 8.0 containing 20 mM CaCl2 and 100 μM l-BAPA substrate. Chymotrypsin activity was tested using the enzyme at 50 nM in 50 mM Tris–HCl buffer pH 8.0 containing 20 mM CaCl2 and 100 μM Suc-AAPF-pNA substrate. The chymotrypsin-like activity of proteasome (170 nM) was measured in 25 mM Tris–HCl pH 7.5 using 50 μM of N-succinyl-Leu–Leu–Val–Tyr-7-amido-4-methylcoumarin substrate. The enzymatic activity was monitored on a Cary 100 Scan (Varian) UV/Vis spectrofluorimeter, equipped with a thermostated cuvette compartment using as excitation wavelength 345 nm and as emission wavelength 445 nm. Protease inhibiting activities were carried out using a fixed amount of enzymes and increasing peptide concentrations (25, 50 and 100 μM). Mixtures were pre-incubated for 30 min at 37 °C before the addition of the substrate, and the enzymatic activities were followed as described above. Experiments were repeated at least three times.Conflict of interest
The authors declare no financial or commercial conflict of interest.Abbreviations
| DMF | Dimethylformamide |
| DCM | Dichloromethane |
| DIEA | N,N-Diisopropylethylamine |
| HBTU | O-Benzotriazole-N,N,N′,N′-tetramethyl-uronium-hexafluoro-phosphate |
| HOBt | N-Hydroxybenzotriazole |
| HPLC | High Performance Liquid Chromatography |
| LC-MS | Liquid Chromatography Mass Spectrometry |
| NHB | Non-Hydrogen Bonded |
| TIS | Tri-isopropylsilane |
| TFA | Trifluoroacetic acid |
Acknowledgements
This work was supported by funds from FIRB MERIT no. RBNE08NKH7_003 and from PON Ricerca e Competitività 2007–2013 (PON01_01602, PON01_02342) to MR. AS is fully supported by FIRB no. RBNE08NKH7_003.References
- Z. Szeltner, D. Rea, T. Juhász, V. Renner, V. Fülöp and L. Polgár, J. Mol. Biol., 2004, 340, 627 CrossRef CAS PubMed
.
- D. Rea and V. Fülöp, Cell Biochem. Biophys., 2006, 44, 349 CrossRef CAS
- K. K. Sharma and B. J. Ortwerth, Eur. J. Biochem., 1993, 216, 631 CrossRef CAS PubMed
- E. A. Brunialti, P. Gatti-Lafranconi and M. Lotti, Biochimie, 2011, 93, 1543 CrossRef CAS PubMed
- J. Perrier, A. Durand, T. Giardina and A. Puigserver, Biochimie, 2005, 87, 673 CrossRef CAS PubMed
- G. Palmieri, P. Bergamo, A. Luini, M. Ruvo, M. Gogliettino, E. Langella, M. Saviano, R. N. Hegde, A. Sandomenico and M. Rossi, PLoS One, 2011, 6, 1 Search PubMed
- K. Shimizu, T. Fujino, K. Ando, M. Hayakawa, H. Yasuda and K. Kikugawa, Biochem. Biophys. Res. Commun., 2003, 304, 766 CrossRef CAS
- A. Scaloni, W. Jones, M. Pospischil, S. Sassa, O. Schneewind, A. M. Popowicz, F. Bossa, S. L. Graziano and J. M. Manning, J. Lab. Clin. Med., 1992, 120, 546 CAS
- R. Erlandsson, F. Boldog, B. Persson, E. R. Zabarovsky, R. L. Allikmets, J. Sümegi, G. Klein and H. Jörnvall, Oncogene, 1991, 6, 1293 CAS
- S. L. Naylor, A. Marshall, C. Hensel, P. F. Martinez, B. Holley and A. Y. Sakaguchi, Genomics, 1989, 4, 355 CrossRef CAS
- M. Yamaguchi, D. Kambayashi, J. Toda, T. Sano, S. Toyoshima and H. Hojo, Biochem. Biophys. Res. Commun., 1999, 263, 139 CrossRef CAS PubMed
- P. Bergamo, E. Cocca, R. Palumbo, M. Gogliettino, M. Rossi and G. Palmieri, PLoS One, 2013 DOI:10.1371/journal.pone.0080900
- A. Sandomenico, A. Russo, G. Palmieri, P. Bergamo, M. Gogliettino, L. Falcigno and M. Ruvo, J. Med. Chem., 2012, 55, 2102 CrossRef CAS PubMed
- A. Adibekian, B. R. Martin, C. Wang, K. L. Hsu, D. A. Bachovchin, S. Niessen, H. Hoover and B. F. Cravatt, Nat. Chem. Biol., 2011, 7, 469 CrossRef CAS PubMed
- V. Celentano, D. Diana, L. De Rosa, A. Romanelli, R. Fattorusso and L. D. D'Andrea, Chem. Commun., 2012, 48, 762 RSC
- S. Ebdrup, L. G. Sorensen, O. H. Olsen and P. Jacobsen, J. Med. Chem., 2004, 47, 400 CrossRef CAS PubMed
- R. Jagasia, J. M. Holub, M. Bollinger, K. Kirshenbaum and M. G. Finn, J. Org. Chem., 2009, 74, 2964 CrossRef CAS PubMed
- C. W. Tornoe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057 CrossRef CAS PubMed
- H. Li, R. Aneja and I. Chaiken, Molecules, 2013, 18, 9797 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra13505a |
| ‡ These authors equally contributed to this work. |
| This journal is © The Royal Society of Chemistry 2015 |