DOI:
10.1039/C4RA13486A
(Paper)
RSC Adv., 2015,
5, 75-84
Deep-blue electroluminescence from nondoped and doped organic light-emitting diodes (OLEDs) based on a new monoaza[6]helicene†
Received
30th October 2014
, Accepted 20th November 2014
First published on 1st December 2014
Abstract
A new organic electroluminescent material, monoaza[6]helicene (1), was successfully prepared via an effective photochemical reaction in about 10 minutes with high productivity. The thermal, photophysical, electrochemical properties, quantum chemical and X-ray structural studies as well as characteristics of organic light-emitting diodes were fully investigated. 1 exhibited excellent solubility and high thermal stability. The first singlet excitation energy of 1 was measured to be around 2.92 eV with a HOMO level of −5.19 eV and a LUMO level of −2.27 eV. Helical molecular geometry of 1 effectively blocked the π-conjugation and decreased the close-packing of molecules. Both the nondoped and doped OLEDs emitted deep-blue light at 444 and 462 nm with CIE coordinates of (0.15, 0.09) and (0.15, 0.10). The doped OLED based on 1 was the first example of helicenes as light emitters that exhibited a brightness of more than 3000 cd m−2 with high color purity and excellent efficiency stability.
Introduction
OLEDs based on small conjugated molecules and polymers have attracted great scientific and technical interest for applications in display and solid-state lighting sources.1–7 As is known, red and green organic emitters capable of sufficient efficiency, color purity, and lifetime have been developed for commercial application, whilst their blue organic counterparts are still in the path towards commercialization.8–13 This results from the following facts: (1) the intrinsic wide band gap for blue emission generally leads to low electron affinity for blue-light-emitting materials, an imbalance in charge transport and low efficiency for blue OLEDs; (2) design and preparation of deep-blue organic emitters endowed with an amorphous state upon thermal evaporation and suppression of π-conjugation is a rather daunting task.14 Blue organic light-emitting materials, which have blue electroluminescence (EL) emission with a Commission Internationale de l'Eclairage (CIE) y-coordinate value of less than 0.15, are of extreme significance to realize practical applications by being utilized to decrease the power consumption of the devices and as hosts for energy transfer to lower-energy fluorescent or phosphorescent dopants to render efficient emission of other colors.15,16 Although there are some problems with many classes of blue organic electroluminescent materials under current investigation, for example, low thermal and morphological stabilities, low device efficiencies, poor color purity, and so on, research efforts remain strong to seek for blue organic light-emitting materials with blue emission of relatively balanced stability, color purity, and efficiency.
According to one of strategies to design deep-blue emitters with both excellent Commission Internationale de l'Eclairage (CIE) coordinates and high efficiencies, introduction of bulky and non-planar side groups to the planar core is usually an effective method to get out of the dilemma, which not only prevents π–π stacking interactions but also leads to forming amorphous films and hindering exciton quenching in high concentration.17,18 Introducing long alkyl substitutions to a suitable helicence is another way to fulfill this daunting task. A helicene is polycyclic aromatic compound with a nonplanar screw-shaped skeleton formed by ortho-fused benzene or other aromatic rings.19–24 On the one hand, helical molecular geometry results in conjugation blocking for wide band gaps and nonplanar twist structures, which reduces molecular aggregations, keeps molecules amorphous, and hinders exciton quenching in high concentration. On the other hand, this geometry is unfavorable to π-electron overlaps, charge carrier transport and exciton generation, even decreases device efficiency. The helical molecule structure of well-designed helicenes could cleverly keep an amorphous state upon thermal evaporation, suppression of π-conjugation and device efficiencies in balance. Furthermore, a helicene as a rigid backbone, often has a good fluorescent property and a high decomposition temperature. Its unique structure and property aroused people to explore possibilities for various applications, such as asymmetric catalysis,25 enantioselective fluorescent sensors,26 chiral discotic liquid-crystalline materials,27 nonlinear optical materials,28 and organic light-emitting materials.29 As for EL materials, carbazole is a popular functional unit in the conjugated systems with a wide band-gap and N–H bond, which can be easily substituted by alkyl substitutions. Introducing alkyl substitutions of suitable length to organic molecules is a key approach not only to ensure the solubility in solvents for solution fabrication of devices,30,31 but also to facilitate the homogeneous film without grain boundaries or pin holes for avoiding current leakage of the device.32 Despite the fact that a carbazole-based helicene as a blue organic emitter was characterized by excellent deep-blue Commission Internationale de l'Eclairage (CIE) chromaticity coordinates of (0.15, 0.10), brightness of 2365 cd m−2, high thermal and film morphological stabilities,29 high performance blue-emitting helicene-based materials are still needed to be explored.
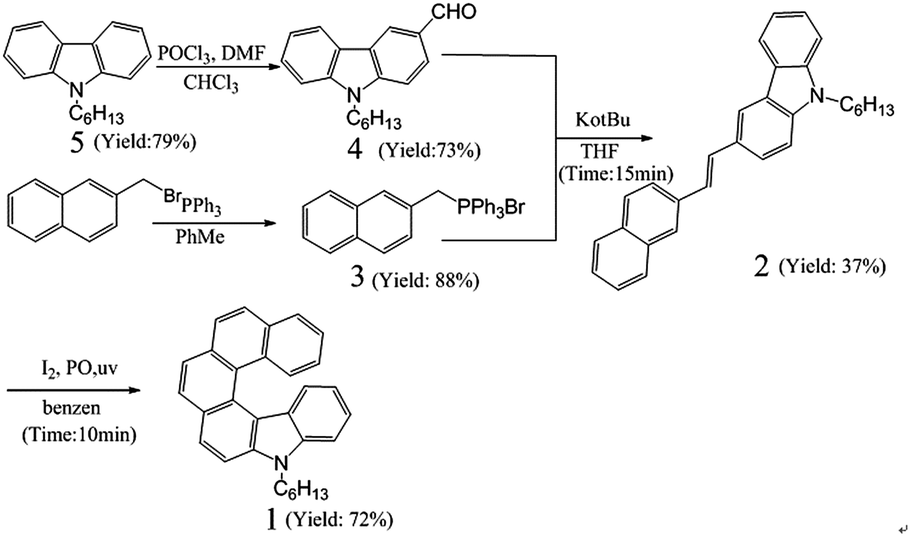
Herein, a novel nitrogenous helicene with six rings, namely 7-hexyl-7-monoaza[6]helicene (1), was designed and synthesized through photocyclization reaction as the key step (Scheme 1 and S1†) in an exceedingly short time (ca. 10 min) with high productivity (72.0%). Such efficient and convenient reaction renders it feasible way to synthesize other carbazole-based helicenes. Compound 1 used as deep-blue nondoped and doped emitters for OLEDs was systematically investigated. It was confirmed that 1 exhibited high solubility in common organic solvents, which overcame the formidable problem of low-solubility in synthesis of long helicenes as highly rigid and annelated structures.33 By slow evaporation from the ethanol and dichloromethane mixed solution, we fortunately acquired the crystals of 1. Moreover, 1 showed excellent thermostability with a decomposition temperature (Td) reaching 297.0 °C and the melting temperature (Tm) of 130.9 °C. Single-crystal structure analysis along with theoretical investigation revealed the non-planar crystal structure of 1 responsible for reducing close-packing arrangement, keeping the molecule in a good amorphous state, and blocking exciton quenching in high concentration. These merits make compound 1 a good candidate for deep-blue emitters. In order to study its electroluminescent behaviors in nondoped and doped devices, a series of devices A and B with multilayer configurations (Fig. 7(a)) were fabricated. Device A has a configuration of ITO/NPB (50 nm)/1 (30 nm)/Bphen (20 nm)/Mg:Ag (150 nm)/Ag (50 nm). The emitting layer of 1 is substituted for that of CBP:10% 1 (30 nm) in device B. Both devices have the same total thickness of organic films (100 nm). The nondoped device A presented saturated blue light with the CIE coordinates of (0.15, 0.09), a maximum current efficiency (CE) of 0.42 cd A−1 (0.11 lm W−1), brightness of 1208.90 cd m−2, and a maximum external quantum efficiency (EQE) of 0.51%, respectively. In contrast, device B with the CIE coordinates of (0.15, 0.10) exhibited high performances featuring with a maximum CE of 0.68 cd A−1 (0.17 lm W−1), brightness of 3245.25 cd m−2, and EQE of 0.76%. Compared with the diaza[7]helicene-based doped OLED, not only a nondoped OLED based on the monoaza[6]helicene was first fabricated, whose key performance parameters such as the maximum current efficiency, maximum power efficiency, and the maximum external quantum efficiency were superior to those of the doped diaza[7]helicene-based OLED, but also the performance of the doped monoaza[6]helicene-based OLED was much higher than that of the doped diaza[7]helicene-based one.29 The new class of helicenes opens new possibilities for utilizing helicenes in deep-blue OLEDs and other fields.
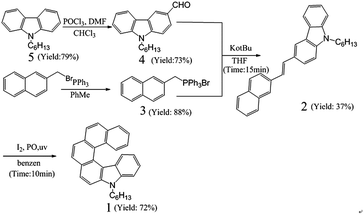 |
| | Scheme 1 The synthetic procedures for compound 1. | |
Experimental section
General information
Reagents were used as purchased without further purification unless otherwise stated. DMF was dried with molecular sieves. THF was refluxed with sodium in the presence of benzophenone.
Photocyclization was performed using a Hanovia high-pressure mercury lamp (500 W). 1H and 13C spectra were recorded on a Bruker Advance 300 spectrometer. FTIR spectrum was recorded on a Thermo-Nicolet NEXUS spectrometer (Thermo-Nicolet Company, USA). HRMS spectra were recorded on a Q-TOF6510 spectrograph (Agilent). X-ray diffraction intensity data were collected at the temperature of 130 K on a Bruker Smart Apex2 CCD area-detector diffractometer equipped with graphite-monochromated Mo Kα radiation (λ = 0.71073 Å). Processing of the intensity data was carried out by using the Bruker SMART routine, and the structure was solved by direct methods by using SIR-97 program and refined by a full-matrix least-squares technique based on F2 with the SHELXL-97 program (ESI†).34,35
TG and DSC curves were obtained on a SDT Q600 V8.0 Differential Scanning Calorimeter under nitrogen atmosphere with heating and cooling rates of 10 °C min−1. Cyclic voltammetry (CV) measurement of compound 1 was run on a CHI660D electrochemical workstation. A conventional three-electrode system with a glassy carbon working electrode, a Pt counter electrode, and an Ag/AgCl reference electrode was employed. 0.1 M [Bu4N][ClO4] CH2Cl2 solution was used as the supporting electrolyte. The experiment was corrected by using the ferrocene/ferrocenium (Fc/Fc+) redox system. UV/Vis absorption spectroscopy was performed on a TU-1800 spectrophotometer (Hitachi). PL spectra were recorded on a Hitachi F-4500 fluorescence spectrophotometer. The neat films were prepared by spin-coating on SiO2 glass slides. Fluorescence quantum yields were calculated by using a quinine sulfate in 0.1 M H2SO4 solution as a reference.36 The PL quantum yields of neat films were measured by an absolute method using the Edinburgh Instruments (FLS920) integrating sphere excited with Xe lamp. DFT and TD-DFT calculations were carried out using Gaussian 09 program.37 The target molecule was assumed to be an isolated molecule in the gas phase.
Device fabrication and measurement
4,4′-Bis(1-naphthyl-N-phenylamino)-biphenyl (NPB) and bathophenanthroline (Bphen) were commercially available. Commerical ITO coated glass with sheet resistance of 7 Ω per square was used as the starting substrates. Prior to device fabrication, indium-tin-oxide (ITO) glass substrates were cleaned sequentially by ultrasonication in organic solvent and de-ionized water and dried for 2 h. After UV-ozone and plasma treatments, the organic layers and cathode layers were sequentially deposited onto the ITO coated glass substrate by vacuum deposition at the rate of 1 Å s−1 in the vacuum of 3–4 × 10−4 Pa. The substrate was patterned with an active area of 3 × 3 mm2. Then organic layers and cathode layers were vacuum evaporated successively. In two types of devices, NPB and Bphen were hole-transport and electron-transport layers, respectively. Compound 1 was utilized as the emitting layer (EML) in device A while 10 wt% 1 in CBP was employed as EML in device B. ITO and Mg:Ag were served as the anode and the cathode, respectively. The current density–voltage–luminescence (J–V–B) of devices was measured with a Keithley 4200 semiconductor characterization system, the electroluminescence (EL) spectra and CIE coordinates were obtained by a Photo Research PR705 spectrophotometer. The external quantum efficiencies were calculated from the corresponding current efficiencies. All the measurements were performed at room temperature under ambient conditions without encapsulation.
Synthesis
9-Hexyl-9H-carbazole (5) was synthesized referring to the reported procedures.38 7-Hexyl-7-monoaza[6]helicene (1) was prepared with reference to our previous method.29
9-Hexyl-9H-carbazole (5). To a mixture of TBAB (3 g, 50 mmol), carbazole (33.4 g, 200 mmol) and NaOH (12 g, 300 mmol) was added acetone (140 mL). The solution was heated to 60 °C and stirred at this temperature for 3 h. After cooling to room temperature, 1-bromohexane (42.6 mL, 300 mmol) in acetone (60 mL) was added dropwise to the solution. The reaction mixture was then stirred at 60 °C for 24 h. After the solvent was removed under reduced pressure, the residue was poured into ice water and stirred vigorously for 2 h. The precipitate was filtered and washed with ethanol for 3 times (3 × 50 mL) to afford compound (3) as a white solid (40 g). Yield: 79%. 1H (300 MHz, CDCl3, δ) 8.11 (s, 1H), 8.08 (s, 1H), 7.48–7.38 (m, 4H), 7.24–7.19 (m, 2H), 4.28 (t, J = 7.2 Hz, 2H), 1.91–1.81 (m, 2H), 1.43–1.22 (m, 6H), 0.86 (t, J = 6.9 Hz, 3H). 13C (75 MHz, CDCl3, δ) 140.45, 125.56, 122.84, 120.33, 118.69, 108.65, 43.10, 31.60, 28.95, 26.99, 22.55, 14.01. HRMS (m/z): 252.1736 [M + H]+ (calcd for C18H21N: 252.1747).
9-Hexyl-9-carbazole-3-carbaldehyde (4). POCl3 (1.17 mL, 12.55 mmol) was added dropwise to DMF (1.13 mL, 14.6 mmol) at 0 °C, and stirred for 30 min under Ar. A solution of 5 (3.145 g, 12.50 mmol) in CHCl3 (7.5 mL) was then added. The reaction mixture was heated to 75 °C and stirred at this temperature. After 16 h, the reaction mixture was poured into ice water under stirring and neutralized by the saturated aqueous NaHCO3. The mixture was extracted by CH2Cl2. The organic layer was washed with water for three times and dried over anhydrous Na2SO4. After the solvent was removed in vacuo, the residue was purified by flash chromatography on silica gel using ethyl acetate–petroleum ether (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20) as the eluent to give a white solid (2.55 g). Yield: 73%. 1H (300 MHz, CDCl3, δ) 10.09 (s, 1H), 8.61 (d, J = 1.2 Hz, 1H), 8.15 (d, J = 7.8 Hz, 1H), 8.00 (dd, J = 8.7 Hz, 1H), 7.56–7.44 (m, 3H), 7.35–7.29 (m, 1H), 4.33 (t, J = 7.5 Hz, 2H), 1.94–1.84 (m, 2H), 1.42–1.28 (m, 6H), 0.86 (t, J = 6.9 Hz, 3H). 13C NMR (75 MHz, CDCl3, δ) 191.71, 144.08, 141.19, 128.54, 127.14, 126.68, 123.96, 123.08, 123.01, 120.73, 120.27, 109.37, 108.92, 43.44, 31.49, 28.88, 26.90, 22.49, 13.94. HRMS (m/z): 280.1652 [M + H]+ (calcd for C19H21NO: 280.1696).
:20) as the eluent to give a white solid (2.55 g). Yield: 73%. 1H (300 MHz, CDCl3, δ) 10.09 (s, 1H), 8.61 (d, J = 1.2 Hz, 1H), 8.15 (d, J = 7.8 Hz, 1H), 8.00 (dd, J = 8.7 Hz, 1H), 7.56–7.44 (m, 3H), 7.35–7.29 (m, 1H), 4.33 (t, J = 7.5 Hz, 2H), 1.94–1.84 (m, 2H), 1.42–1.28 (m, 6H), 0.86 (t, J = 6.9 Hz, 3H). 13C NMR (75 MHz, CDCl3, δ) 191.71, 144.08, 141.19, 128.54, 127.14, 126.68, 123.96, 123.08, 123.01, 120.73, 120.27, 109.37, 108.92, 43.44, 31.49, 28.88, 26.90, 22.49, 13.94. HRMS (m/z): 280.1652 [M + H]+ (calcd for C19H21NO: 280.1696).
Bromo(naphthalen-2-ylmethyl)triphenylphosphorane (3). A mixture of 2-bromine methyl naphthalene (1 g, 4.5 mmol) and PPh3 (1.31 g, 5 mmol) was dissolved in toluene (90 mL). The solution was heated to 90 °C and stirred at this temperature for 12 h. The resulting precipitate was filtered and washed with petroleum ether for 3 times (3 × 100 mL) to give a white solid (1.92 g). Yield: 88%. 1H (300 MHz, CDCl3, δ) 7.78–7.67 (m, 10H), 7.62–7.51 (m, 9H), 7.44–7.35 (m, 2H), 7.16 (d, J = 8.4 Hz, 1H), 5.58 (s, 1H), 5.53 (s, 1H). 13C (75 MHz, CDCl3, δ) 134.95, 134.91, 134.51, 134.38, 132.95, 132.90, 132.66, 132.62, 131.27, 131.17, 130.19, 130.03, 128.64, 128.59, 128.38, 128.35, 127.85, 127.54, 127.52, 126.54, 126.35, 124.55, 124.43, 118.42, 117.28, 53.44, 31.33, 30.71. HRMS (m/z): 403.3373 [M − Br]+ (calcd for C29H24BrP: 403.1616).
(E)-9-Hexyl-3-(2-(naphthalen-2-yl)vinyl)-9H-carbazole (2). Compound (3) (4.5 g, 9.3 mmol) was dissolved in dry THF (20 mL) under Ar and the solution was cooled to 0 °C. t-BuOK (2 g, 20.4 mmol) was added and stirred for 30 min. A solution of compound 4 (3.5 g, 10 mmol) in dry THF (20 mL) was added dropwise into the reaction mixture. After addition, the mixture was stirred for 1 h. A few drops of water were added to quench the reaction. The mixture was extracted by CH2Cl2 (3 × 100 mL). The organic layer was washed with water for three times (3 × 100 mL) and dried over anhydrous Na2SO4. The solvent was removed in vacuo, the residue was purified by flash chromatography on silica gel using dichloromethane–petroleum ether (1:10) as the eluent to afford a white solid (1.4 g). Yield: 37%. 1H NMR (300 MHz, CDCl3, δ) 8.28 (d, J = 1.2 Hz, 1H), 8.14 (d, J = 7.8 Hz, 1H), 7.89 (s, 1H), 7.86–7.78 (m, 4H), 7.73 (dd, J = 8.7 Hz, 1H), 7.50–7.34 (m, 7H), 7.29–7.23 (m, 1H), 4.31 (t, J = 7.2 Hz, 2H), 1.89 (m, 2H), 1.38–1.22 (m, 6H), 0.87 (t, J = 6.9 Hz, 3H) 13C NMR (75 MHz, CDCl3, δ) 140.89, 140.32, 135.53, 133.87, 132.80, 130.07, 128.53, 128.22, 127.89, 127.69, 126.24, 126.08, 125.91, 125.81, 125.55, 124.46, 123.59, 123.25, 122.89, 120.43, 119.01, 118.73, 108.92, 108.88, 43.24, 31.58, 28.98, 26.97, 22.54, 13.99. HRMS (m/z): 404.2379 [M + H]+ (calcd for C30H29N: 404.2373).
7-Hexyl-7-monoaza[6]helicene (1). Argon was bubbled through a solution of compound (2) (0.182 g, 0.45 mmol) in benzene (500 mL) with stirring for 30 min. After I2 (0.117 g, 0.46 mmol) and propylene oxide (18 mL, 258 mmol) were added into the solution, the reaction mixture was irradiated by a 500 W high pressure mercury lamp through a quartz filter. The reaction was stopped 10 min later. The residue was dissolved in CH2Cl2 (20 mL) when the solvent had been removed in vacuo. The solution was then washed with aqueous Na2S2O3 (60 mL, 15%) and water sequentially. The organic layer was purified by flash chromatography on silica gel using dichloromethane–petroleum ether (1:10) as the eluent to give a yellow solid (0.13 g). Yield: 72.0%. 1H NMR (300 MHz, CDCl3, δ) 9.35 (d, J = 8.4 Hz, 1H), 9.07 (s, 1H), 8.70 (s, 1H), 8.29 (d, J = 7.8 Hz, 1H), 8.10–8.05 (m, 2H), 7.89 (q, J = 8.4 Hz, 2H), 7.73–7.54 (m, 4H), 7.46 (d, J = 8.1 Hz, 1H), 7.33–7.28 (m, 1H), 4.44 (t, J = 7.2 Hz, 2H), 2.02 (m, 2H), 1.58–1.25 (m, 6H), 0.87 (t, J = 6.9 Hz, 3H). 13C NMR (75 MHz, CDCl3, δ) 142.76, 140.65, 133.39, 130.90, 130.76, 129.06, 128.71, 128.41, 127.66, 127.22, 127.07, 127.03, 126.94, 125.87, 125.39, 123.75, 123.63, 122.62, 121.07, 119.35, 118.90, 108.51, 105.50, 43.37, 31.71, 28.90, 27.23, 22.62, 14.02 FTIR (Smart iTR diamond ATR accessory) (ν/cm−1): 3050 (Ar-H); 2955, 2923, 2871, 2856 (C–H, Aliph); 1606, 1492, 1470, (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, Ar); 1365, 1322, (C–N). HRMS (m/z): 402.2214 [M + H]+ (calcd for C30H27N: 402.2216).
C, Ar); 1365, 1322, (C–N). HRMS (m/z): 402.2214 [M + H]+ (calcd for C30H27N: 402.2216).
Results and discussion
Synthesis and characterization
The synthetic procedures for compound 1 were illustrated in Scheme 1. Compound 5 with DMF and POCl3 first underwent Vilsmeier–Hack reaction to give 4, and 2-bromine methyl naphthalene reacted with PPh3 to achieve compound 3. Then, compound 3 experienced Wittig olefination reaction with t-BuOK, compound 4 in THF under argon atmosphere to obtain compound 2. Finally, the target compound 1 was successfully achieved using compound 2, I2 and propylene oxide in benzene solvent under irradiation of a Hanovia high-pressure mercury lamp. The compounds were verified by 1H NMR, 13C NMR, mass spectroscopy and single-crystal X-ray diffraction pattern. Detailed synthetic procedures and characterization of the compounds were described in the Experimental section.
Convenient synthesis and satisfactory product yields made 1 harvested on a large scale. Especially, compound 1 was acquired in the time of around 10 min with high productivity of 72.0%, which provided an efficient and simple route to synthesize other helicenes. Moreover, compound 1 was readily soluble in common organic solvents, such as petroleum ether, n-hexane, toluene, dichloromethane, trichloromethane, THF, methyl alcohol, and acetonitrile due to existing a hexyl substitution and the noncoplanar helical structure. Nonplanar twist structure and long alkyl substitutions not only overcame formidable problems in synthesizing highly rigid and annelated helicenes, but also facilitated homogeneous film and fabrication of devices by solution-processed method.
Structural characterization
Slow evaporation of 1 from dichloromethane–ethanol mixed solution gave pale yellow plate crystals (Fig. S1†). The crystal structure of 1 was elucidated by single-crystal X-ray analysis. Fig. 1 showed the ORTEP drawing and crystal packing structure. The crystal data and structural parameters are summarized in Table 1. The molecular geometry of compound 1 (Fig. 1(a)) comprising carbazole and phenanthrene moieties possessed long and short characteristic C–C bonds.39 C–C bonds with an average of 1.428(4) Å in the inner helical rim of the Fjord region, namely C20–C15 (1.401(3) Å), C15–C8 (1.434(3) Å), C8–C9 (1.423(4) Å), C9–C10 (1.445(4) Å), C10–C11(1.459(4) Å), C11–C27 (1.401(4) Å) increased by 0.033 Å relative to the average bond-length 1.395(3) Å of those in aromatic compounds. In contrast, their counterparts with the average of 1.343(5) Å in the peripheral six-member rings, that is C17–C18 (1.368(4) Å), C30–C29 (1.331(5) Å), C22–C23 (1.342(5) Å), C25–C26 (1.331(5) Å), C13–C14 (1.344(5) Å), decreased by 0.052 Å compared to the average bond-length of those in aromatic compounds. These longer and shorter bonds were resulted from intramolecular torsion. It is worth noting that lengths of C30–C29, C22–C23 and C25–C26 bonds in the peripheral six-member rings were significantly shorter than those of its counterparts C8–C9, C9–C10 and C10–C11, which demonstrated more twisting to reduce steric hindrance between the naphthalene and carbazole units.
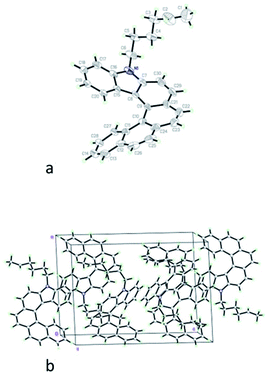 |
| | Fig. 1 (a) Molecular structure of 1 with 30% probability ellipsoids. (b) The crystal-packing diagram along the crystallographic b axis. | |
Table 1 Crystallographic data and structure refinement for 1
| 1 |
| R1 = ∑∥Fo| − |Fc∥. wR2 = [∑[w(Fo2 − Fc2)2]/∑[w(Fo2)2]]1/2; w = 1/[σ2(Fo2) + (0.095P)2]; P = [max(Fo2, 0) + 2Fc2]/3 (based on reflections with Fo2 > 2σ(Fo2)). |
| Empirical formula |
C30H27N |
| Formula weight |
401.54 |
| Crystal system |
Monoclinic |
| Z, space group |
4, P21/c |
| a (Å) |
12.919(4) |
| b (Å) |
9.577(3) |
| c (Å) |
17.878(6) |
| β (°) |
93.149(5) |
| Volume (Å3) |
2208.7(13) |
| Max. and min. transmission |
0.7456 and 0.5926 |
| Calculated density (g cm−3) |
1.168 |
| Absorption coefficient (mm−1) |
0.067 |
| F (000) |
828 |
| Crystal size (mm) |
0.80 × 0.21 × 0.05 |
| 2θmax (°) (completeness) |
55.24 (97.6%) |
| Reflections collected/unique |
13043/5003 [R(int) = 0.0627] |
| Data/restraints/parameters |
5003/0/281 |
| Goodness of fit, F2 |
1.233 |
| Final R indices [I > 2σ(I)] |
R1a = 0.0834, wR2b = 0.2129 |
| R indices (all data) |
R1a = 0.1457, wR2b = 0.2466 |
| Largest diff. peak/hole (e Å−3) |
0.379 and −0.272 |
The dihedral angle between the planar carbazole and naphthalene segments were 45.0°. In the crystal packing structure of 1 (Fig. 1(b)), both C–H⋯π edge-to-face intermolecular interactions and π–π face-to-face intermolecular interactions between carbazole rings were observed. The intermolecular distance of C–H⋯π edge-to-face between C16–H29 was 2.834 Å and that of the π–π face-to-face between C16–C17 was found to be 3.358 Å, which might cause emission peaks of PL and EL spectra in film-state red-shifted compared to those in solution-state. Despite of these weak intermolecular interactions. The non-coplanar fusion structure of 1 and steric effect of long substituted alkyl chain still reduced close-packing arrangement and kept the molecule in a good amorphous state.
Thermal properties
Thermal properties of 1 in crystalline-state were investigated by thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) with heating and cooling rates of 10 °C min−1 under N2. DSC measurements were performed from 20.0 to 150.0 °C for the first cycle and from 20 to 200 °C for the second cycle. During the first heating process, an endothermic peak was found at a melting point (Tm) of 130.9 °C. When the sample was heated again, a glass-transition revealed at 101.0 °C. As shown in Fig. 2, decomposition temperature (Td, defined as temperature at which there is a 5% weight loss), was measured to be 297.0 °C, indicating high thermal stability of 1. Note that a high Tg value is a great asset to OLED applications, as Joule heating occurs under typical operating conditions.40 Furthermore, The temperature of the large span of ca. 166.1 °C between Tm and Td would benefit vacuum thermal sublimation for OLED fabrication.
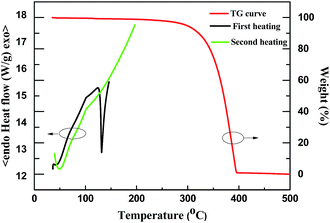 |
| | Fig. 2 TG and DSC curves of 1 recorded under nitrogen at heating and cooling rates of 10 °C min−1. | |
Electrochemical properties
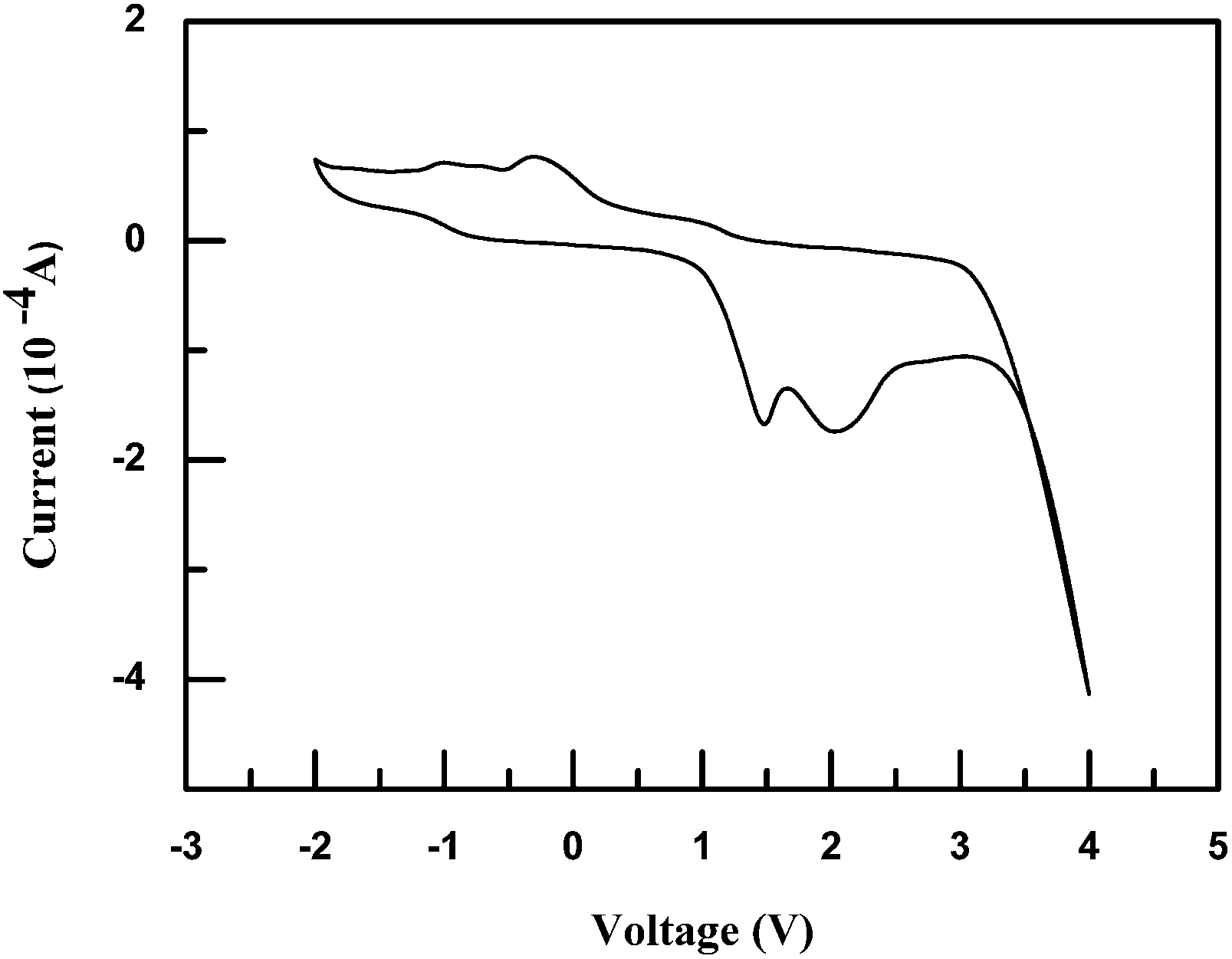
Compound 1 was dissolved in anhydrous and deaerated dichloromethane at 10−4 M. A glassy carbon electrode, a platinum electrode and a saturated Ag/AgCl electrode were employed as working, auxiliary and reference electrodes, respectively. Ferrocene was measured at the beginning and end of the electrochemical measurements to serve as a reference.41 The cyclic voltammogram of compound 1 in anodic and cathodic scans was shown in Fig. 3. In this arrangement, the half-wave potential of Fc/Fc+ (E1/2,FOC) was 0.587 V versus Ag/AgCl. Compound 1 was found to undergo two consecutive oxidative processes. The oxidation peaks of compound 1 were at 1.493 and 2.024 V with its onset oxidation potential of 0.973 V while two reducible peaks were found at −0.306 and at −0.998 V. The redox process was thought to be quasi-reversible. This was attributed to the following facts: (1) shapes of two peaks in the reducible part and those in the oxidative part were asymmetric; (2) two quasi-reversible redox couples, that is, the peak at −0.306 V to that at 1.493 V and the peak at −0.998 V to that at 2.024 V, occurred.
 |
| | Fig. 3 CV of compound 1 in CH2Cl2 (0.1 mol L−1 Bu4NClO4) at a scanning rate of 200 mV s−1. | |
From the onset oxidation potential in the cyclic voltammogram, the highest occupied molecular orbital (HOMO) level has been calculated according to the equation HOMO = −e(Eonset(ox) − E1/2,FOC) − 4.8 eV,42 which is equivalent to ionization potential (IP). Thus, the HOMO level of compound 1 was estimated to be −5.19 eV. Correspondingly, the energy of the LUMO level, as calculated by subtraction of the optical band gap (2.92 eV) from the HOMO level, was −2.27 eV.
Optical properties and quantum chemical calculations
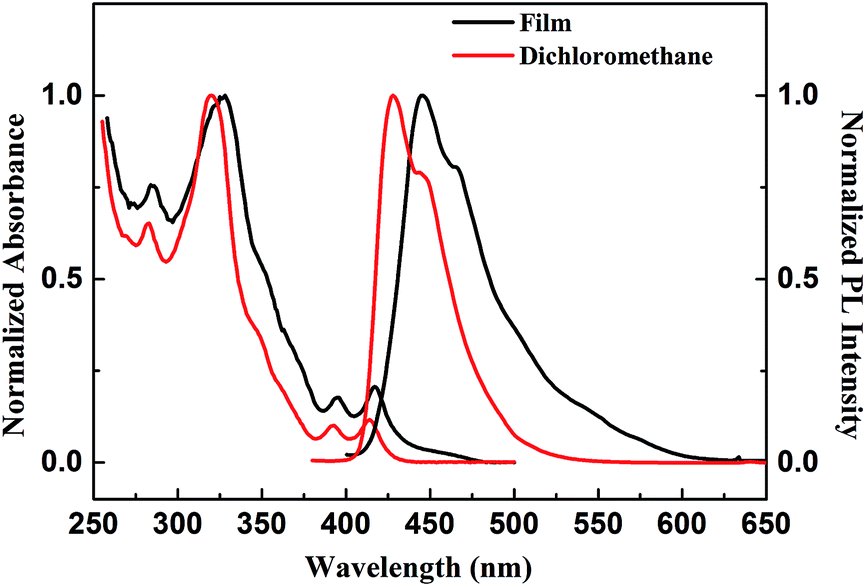
The solution-state and film-state absorption and fluorescence spectra for 1 were recorded and were shown in Fig. 4 and 5. Compound 1 had one absorption maximum peak at 320, one medium peak at 282 nm, one broad shoulder at 347 nm, and two minima at 393 and 414 nm in dilute dichloromethane solution. The absorption behaviors of compound 1 in dilute toluene, THF, trichloromethane, acetonitrile, and methanol solutions resembled those of in dichloromethane with absorption maxima of 315–320 nm, medium peaks within 280–283 nm, shoulders at 343–348 nm and minimum peaks around 393 and 414 nm. A solvent change from toluene to methanol caused small peak shifts of 1–5 nm in its all absorption bands. Thus, compound 1 exhibited almost solvent independence of UV/Vis absorption spectra. Compared with solution spectra, the corresponding peaks and shoulders of thin film showed slight bathochromic shifts by about 2–8 nm, indicating that the twisted and non-coplanar geometry and a long hexyl substitution attached to the helical core could decrease close-packing in the solid state. More than 10 nm hypsochromic shifts of absorption bands around 320 and 280 nm were observed for compound 1 compared to the corresponding ones of its linear coplanar precursor 2 (Fig. S19†). This suggested that the twisted and non-coplanar geometry could decrease π conjugation of 1. The LUMO/HOMO energy gap was approximately estimated to be 2.92 eV from the absorption edge of 425 nm,43 corresponding to the singlet–singlet S0 → S1 0–0 transition.44,45
 |
| | Fig. 4 Absorption and PL spectra of 1 in different solutions (1 × 10−5 mol L−1). | |
 |
| | Fig. 5 Normalized absorption and PL spectra in dichloromethane and in thin-film. | |
As shown in Fig. 4, compound 1 exhibited deep-blue emissions with emission peaks ranging from 425 to 428 nm and shoulders at around 444 nm on going from toluene to methanol as solvent polarity increased. Its emission at around λmax = 426 nm was red-shifted only by 164.9 cm−1 (Δλ = 3 nm) as the solvent polarity increased. The small stokes shift of about 680.4 cm−1 (Δλ = 12 nm) as well as solvent-polarity independence of absorption and emission spectra indicated that the molecule was almost nonpolar and its dipole moments and conformation remained unchanged on electronic transition.46 These favorable properties of nonpolar 1 would facilitate stability of blue-light emitting and improvement of device efficiencies. The corresponding emission peaks and shoulder of the thin film underwent a bathochromic shift by about 19 nm. Compared to fluorescent behaviors of 1, compound 2 was solvent-polarity dependence of emission spectra with emission peaks ranging from 404 to 431 nm as solvent-polarity increased (Fig. S19†). PL quantum efficiencies of compounds 1 and 2 in different solvents and in films were also measured (see ESI Tables S1 and S2†). PL quantum yields of 18–32% in solution and 20% in neat films for compound 2 are superior to those of 17% to 21% in solutions and about 4% in neat films for compound 1. Absorption at long-wavelength bands ranging from 400 nm to 425 nm, which generated intermolecular energy transfer upon excitation at 320 nm, was believed to be one important reason for moderate quantum yields for 1.
To perceive the electronic structures of 1, quantum chemical calculations were carried out using the Gaussian 09 program.37 The molecular geometry was first optimized with density functional theory (DFT) by the B3LYP function with 6-31G(d) basis sets. The optimized geometry and electron density distribution of the frontier molecular orbitals were shown in Fig. S18.† Then, time-dependent density functional theory (TD-DFT) calculation at the b3lyp/6-31+g(d) level was performed and vertical electronic transitions for 1 were obtained. The transition energies, oscillator strength, and assignments for the most relevant singlet excited states were listed in Table S3.† The UV-Vis data were calculated by the Swizard program, revision 4.6,47,48 using the Gaussian model. The half-bandwidths, D1/2,I, were taken to be equal to 3000 cm−1, and the simulated spectrum reproducing the experimental data was displayed in Fig. 6. The result indicated that the maximum peak at 328 nm (30488 cm−1) were mainly attributed to the HOMO → LUMO+1 transition, while minima at 365.6 (HOMO−1 → LUMO transition) and 390.5 nm (HOMO → LUMO transition) with the oscillator strength of 0.0402 and 0.0421 were characteristic of forbidden transitions, which was unfavorable for efficient fluorescence. The medium absorptions at 296.7 nm (33704 cm−1) and 282.9 nm (35348 cm−1) corresponded to the HOMO−3 → LUMO and HOMO−2 → LUMO+1 electronic transitions, respectively.
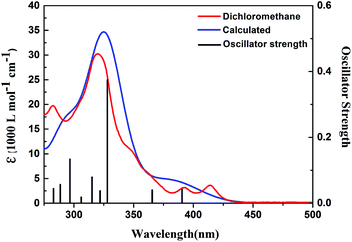 |
| | Fig. 6 TD-DFT calculated absorption spectrum of compound 1 compared with the experimental absorption spectrum (in CH2Cl2 10−5 mol L−1). | |
Electroluminescence properties
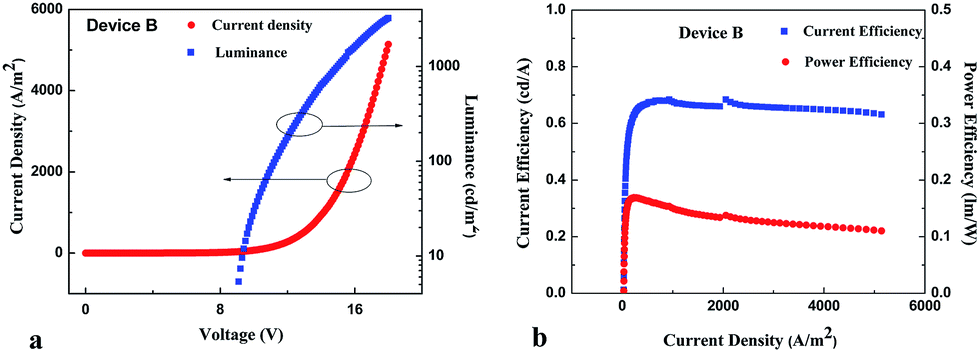
To fully investigate electroluminescence (EL) properties of the novel monoaza[6]helicene, a series of devices A and B with multilayer configurations were fabricated. Device A had a configuration of ITO/NPB (50 nm)/1 (30 nm)/Bphen (20 nm)/Mg:Ag (150 nm)/Ag (50 nm) and device B had the similar structure of ITO/NPB (50 nm)/CBP:10% 1 (30 nm)/Bphen (20 nm)/Mg:Ag (150 nm)/Ag (50 nm). The device configurations and the energy diagrams were shown in Fig. 7. Key device performance parameters were summarized in Table 2.
 |
| | Fig. 7 (a) The device configurations: ITO/NPB (50 nm)/1 or CBP:10% 1 (30 nm)/Bphen (20 nm)/Mg:Ag (150 nm)/Ag (50 nm). (b) Energy-level diagram of the OLED components (relative to the vacuum energy level). | |
Table 2 EL properties of devices A and B for compound 1
| Device no. |
Vonseta (V) |
Bmaxb (cd m−2) |
ηc,maxc (cd A−1) |
ηp,maxd (lm W−1) |
CIEe (x, y) |
λmaxe (nm) |
ηext,maxf (%) |
| Vonset: turn-on voltages were recorded at brightness of 1 cd m−2. Bmax: maximum brightness values were measured at 15 V and 18 V. ηc,max: maximum current efficiencies were measured at 11.7 V and 14.0 V. ηp,max: maximum power efficiencies were measured at 11.7 and 14.0 V. The CIE coordinates and λmax were obtained at the voltage of 8 V. ηext,max: maximum external quantum efficiencies were calculated from corresponding current efficiencies. |
| A |
5.4 |
1209 |
0.42 |
0.11 |
(0.15, 0.09) |
444 |
0.51 |
| B |
5.1 |
3245 |
0.68 |
0.17 |
(0.15, 0.10) |
462 |
0.76 |
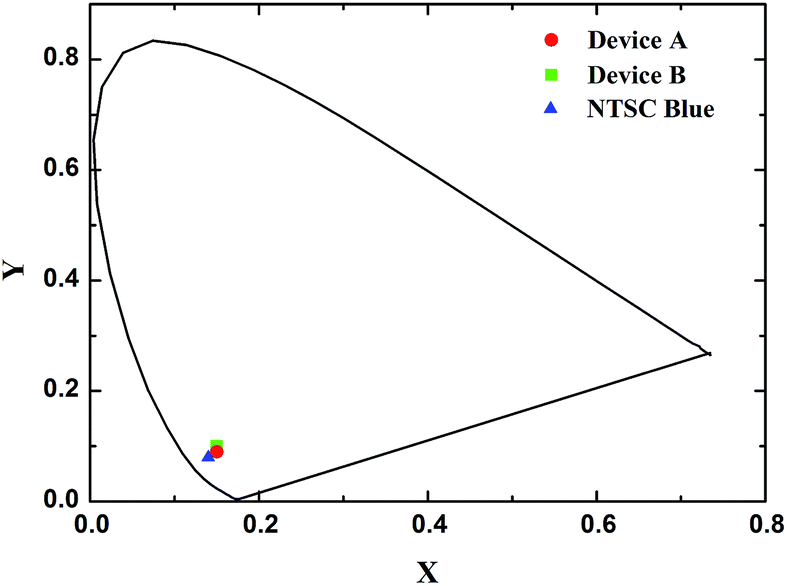
Fig. 8 depicted EL spectra of devices A and B at the driving voltage of 8 V and PL spectra of compound 1 in dichloromethane solution and solid state film. Device A emitted deep blue light of the CIE coordinate (0.15, 0.09) with a dominant peak at 444 nm and a shoulder peak at 462 nm while Device B emitted light of the CIE coordinate (0.15, 0.10) with the same two peaks except for the inverse peak intensity (Fig. 9). As for the difference of the shoulder peaks between device A and B in EL spectra, we looked up literatures about this subject. According to the conclusion drawn from these literatures,49,50 we thought that the difference of the shoulder peaks between device A and B in EL spectra was attributed to the fact the structural distortion that occurred in the excited state relative to the ground state in device B was greater than that in device A. The structural distortion that occurred in the excited state relative to the ground state indicated the degree of vibrational coupling between the ground and excited state (Huang-Rhys factor), which could be estimated by measuring the height of the peak at 462 nm divided by the height of the peak at 444 nm. The EL spectral shapes of the devices were almost the same as those in solid state films and in solution. However, corresponding peaks of solution spectrum were blue-shifted by ca. 16 nm. Moreover, no CBP emission was observed in the spectrum of device B as the blue emission of CBP was centered at λmax = 400 nm.51,52 This indicated that both the conversion of CBP transition states to those of 1 by intermolecular charge transfer and transfer of the energy from the host CBP to the guest 1 were very rapid relative to generation of CBP light emission.
 |
| | Fig. 8 The EL spectra of devices A and B measured at 8 V compared with the PL spectra in dichloromethane and in thin-film. | |
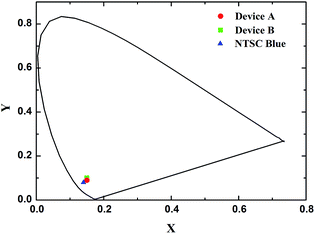 |
| | Fig. 9 CIE coordinates of devices A and B along with the NTSC blue. | |
Current density–voltage–luminance (J–V–L) characteristics of devices A and B were shown in Fig. 10 and 11. NPB had the HOMO energy level at −5.43 eV, it meant that the hole-injection barrier of 0.73 eV at the ITO/NPB junction, and that of 0.77 eV at NPB/CBP junction existed. Similarly, the electron injection barriers at the 1/Bphen, CBP/Bphen and Bphen/Mg:Ag junctions are determined to be 0.63, 0.40, and 0.8 eV, respectively since the LUMO energy level of Bphen was −2.90 eV. These injection barriers accounted for slightly high turn-on voltages of 5.1 V for device A and 5.4 V for device B. In addition, since the electron injection barrier at the CBP/Bphen junction was lower than that at the 1/Bphen junction, this would result in large electron injection efficiency in device B. Thus, relatively balanced charge transport occurred in device B. This might also be responsible for high performance of device B. The device A attained a maximum brightness of 1208.90 cd m−2, maximum current efficiency of 0.42 cd A−1, and power efficiency of 0.11 lm W−1 and external quantum efficiency (EQE) of 0.51%, whose performance parameters were superior or comparable in some aspects to those of carbazole-based EL materials.53–55 In contrast, the device B exhibited high performance featuring a maximum CE of 0.68 cd A−1 (0.17 lm W−1), brightness of 3245.25 cd m−2, and EQE of 0.76%. The performance of device B was much higher than that of device A. Rapid transfer of the energy from the host CBP to the guest 1 might mainly account for great improvements of device B. In addition, considering the concentration of 1 in device B was much lower than that in device A, low aggregate quenching of device B due to low doping concentration was also believed to likely contribute to the dramatic increase of device efficiencies.
 |
| | Fig. 10 (a) Current density (J)–voltage (V)–brightness (b) characteristics of the device A. (b) Current efficiency–current density–power efficiency of device A. | |
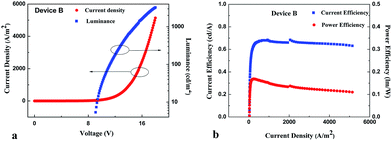 |
| | Fig. 11 (a) Current density (J)–voltage (V)–brightness (b) characteristics of device B. (b) Current efficiency–current density–power efficiency of device B. | |
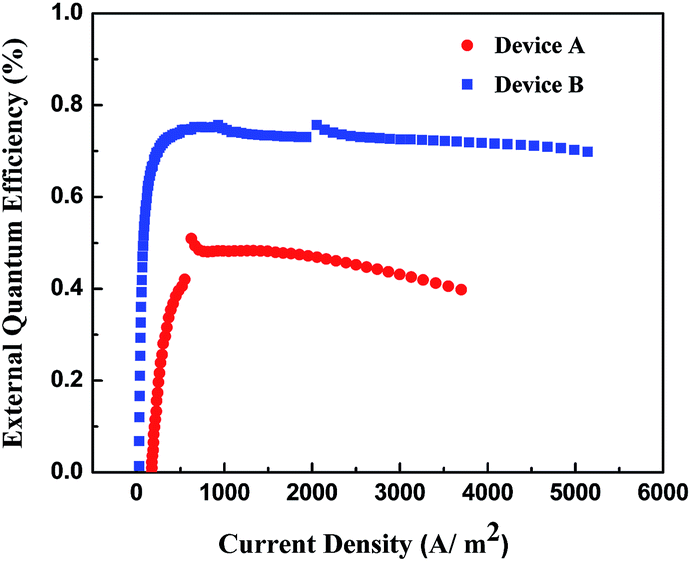
It is worth noting that EQEs of both devices remained almost unchanged except for initial stages as shown in Fig. 12. Even at the highest brightness, external quantum efficiencies were still 0.70% with low roll-off of 7.76% for device B. The low efficiency roll-off at high luminance for device B might be attributed to the small dipole moment of 1.56 Though device A exhibited higher efficiency roll-off than device B, the EQE of device A was still as high as 0.41% (3698.9 A m−2). The performance of device B ranked among the best in either nondoped or doped helicene-based OLEDs.29,57
 |
| | Fig. 12 External quantum efficiencies of devices A and B. | |
Conclusions
In summary, a new carbazole-based 7-hexyl-7-monoaza[6]helicene (1) containing carbazole and phenanthrene moieties, was successfully synthesized by an effective photochemical synthesis method and characterized by 1H NMR, 13C NMR, MS (MALDI-TOF) spectra, and X-ray diffraction pattern. Its thermal, photophysical, electrochemical and electroluminescent behaviors, along with single-crystal structure and electronic structure analyses were also studied. The investigation indicated compound 1 possessed excellent solubility and high thermal stability. Absorption and PL spectra of 1 exhibited solvent-polarity independence. Single-crystal structure analysis along with theoretical investigation revealed helical molecular geometry of 1 prevented the molecules from close-packing in solid state and kept the molecule in a good amorphous state, which could optimize properties of the OLEDs. The performance of OLED using 1 as the dopant emitter with the configuration of ITO/NPB (50 nm)/CBP:10% 1 (30 nm)/Bphen (20 nm)/Mg:Ag (150 nm)/Ag (50 nm) was superior to that of using 1 as the nondoped emitter. This was attributed to its rapid transfer of the energy from the host to the guest and low aggregate quenching of doped device. The doped device achieved a maximum CE of 0.68 cd A−1 (0.17 lm W−1), brightness of 3245.25 cd m−2, and EQE of 0.76% with the CIE coordinates of (0.15, 0.10). Moreover, the doped device still exhibited low efficiency roll-off at the highest brightness. The new class of carbazole-based helicenes was promising for practical applications in optoelectronics and other fields.
Acknowledgements
The research is supported by the National Natural Science Foundation of China (Grant nos 50990061, 51372143, 21373125) and the Natural Science Foundation of Shandong Province (no. ZR2010EM017).
Notes and references
- C. W. Tang and S. A. Vanslyke, Appl. Phys. Lett., 1987, 51, 913–915 CrossRef CAS PubMed.
- H. J. Bolink, F. De Angelis, E. Baranoff, C. Klein, S. Fantacci, E. Coronado, M. Sessolo, K. Kalyanasundaram, M. Gratzel and M. K. Nazeeruddin, Chem. Commun., 2009, 4672–4674 RSC.
- B. Minaev, G. Baryshnikov and H. Agren, Phys. Chem. Chem. Phys., 2014, 16, 1719–1758 RSC.
- H. Wu, L. Ying, W. Yang and Y. Cao, Chem. Soc. Rev., 2009, 38, 3391–3400 RSC.
- K. T. Kamtekar, A. P. Monkman and M. R. Bryce, Adv. Mater., 2010, 22, 572–582 CrossRef CAS PubMed.
- L. Duan, J. Qiao, Y. Sun and Y. Qiu, Adv. Mater., 2011, 23, 1137–1144 CrossRef CAS PubMed.
- G. M. Farinola and R. Ragni, Chem. Soc. Rev., 2011, 40, 3467–3482 RSC.
- P. Wang, Z. Hong, Z. Xie, S. Tong, O. Wong, C. S. Lee, N. Wong, L. Hung and S. Lee, Chem. Commun., 2003, 1664–1665 RSC.
- T. Tsuzuki and S. Tokito, Adv. Mater., 2007, 19, 276–280 CrossRef CAS.
- M. Cocchi, D. Virgili, V. Fattori, D. L. Rochester and J. A. G. Williams, Adv. Funct. Mater., 2007, 17, 285–289 CrossRef CAS.
- M. Zhu and C. Yang, Chem. Soc. Rev., 2013, 42, 4963–4976 RSC.
- C. Liu, Q. Fu, Y. Zou, C. Yang, D. Ma and J. Qin, Chem. Mater., 2014, 26, 3074–3083 CrossRef CAS.
- J. M. Hancock, A. P. Gifford, C. J. Tonzola and S. A. Jenekhe, J. Phys. Chem. C, 2007, 111, 6875–6882 CAS.
- M. T. Lee, C. H. Liao, C. H. Tsai and C. H. Chen, Adv. Mater., 2005, 17, 2493–2497 CrossRef CAS.
- C. H. Chien, C. K. Chen, F. M. Hsu, C. F. Shu, P. T. Chou and C. H. Lai, Adv. Funct. Mater., 2009, 19, 560–566 CrossRef CAS.
- S. Gong, Y. Chen, J. Luo, C. Yang, C. Zhong, J. Qin and D. Ma, Adv. Funct. Mater., 2011, 21, 1168–1178 CrossRef CAS.
- P. I. Shih, C. Y. Chuang, C. H. Chien, E. W. G. Diau and C. F. Shu, Adv. Funct. Mater., 2007, 17, 3141–3146 CrossRef CAS.
- K. Danel, T. H. Huang, J. T. Lin, Y. T. Tao and C. H. Chuen, Chem. Mater., 2002, 14, 3860–3865 CrossRef CAS.
- M. Gingras, Chem. Soc. Rev., 2013, 42, 968–1006 RSC.
- M. Gingras, G. Felix and R. Peresutti, Chem. Soc. Rev., 2013, 42, 1007–1050 RSC.
- T. J. Katz, Angew. Chem., 2000, 112, 1997–1999 (Angew. Chem., Int. Ed., 2000, 39, 1921–1923) CrossRef.
- A. Urbano, Angew. Chem., 2003, 115, 4116–4119 (Angew. Chem., Int. Ed., 2003, 42, 3986–3989) CrossRef.
- R. H. Martin, Angew. Chem., 1974, 86, 727–738 (Angew. Chem., Int. Ed. Engl., 1974
, 13, 649–660) CrossRef CAS.
- A. Rajca and M. Miyasaka, Functional Organic Materials, ed. T. J. J. M ller and U. H. F. Bunz, Wiley-VCH, Weinheim, 2007, pp. 543–577 Search PubMed.
- M. T. Reetz, E. W. Beuttenmiiller and R. Goddard, Tetrahedron Lett., 1997, 38, 3211–3214 CrossRef CAS.
- M. T. Reetz and S. Sostmann, Tetrahedron, 2001, 57, 2515–2520 CrossRef CAS.
- T. Verbiest, S. Sioncke, A. Persoons, L. Vyklicky and T. J. Katz, Angew. Chem., 2002, 114, 4038–4040 (Angew. Chem., Int. Ed., 2002, 41, 3882–3884) CrossRef.
- A. Bossi, E. Licandro, S. Maiorana, C. Rigamonti, S. Righetto, G. R. Stephenson, M. Spassova, E. Botek and B. Champagne, J. Phys. Chem. C, 2008, 112, 7900–7907 CAS.
- L. Shi, Z. Liu, G. Dong, L. Duan, Y. Qiu, J. Jia, W. Guo, D. Zhao, D. Cui and X. Tao, Chem.–Eur. J., 2012, 18, 8092–8099 CrossRef CAS PubMed.
- P. Sonar, M. S. Soh, Y. H. Cheng, J. T. Henssler and A. Sellinger, Org. Lett., 2010, 12, 3292–3295 CrossRef CAS PubMed.
- M. Zhang, S. Xue, W. Dong, Q. Wang, T. Fei, C. Gu and Y. Ma, Chem. Commun., 2010, 46, 3923–3925 RSC.
- M. Zhu, T. Ye, C. G. Li, X. Cao, C. Zhong, D. Ma, J. Qin and C. Yang, J. Phys. Chem. C, 2011, 115, 17965–17972 CAS.
- A. Rajca, M. Pink, S. Xiao, M. Miyasaka, S. Rajca, K. Das and K. Plessel, J. Org. Chem., 2009, 74, 7504–7513 CrossRef CAS PubMed.
- A. Altomare, M. C. Burla, M. Camalli, G. L. Cascarano, C. Giacovazzo, A. Guagliardi, A. G. G. Moliterni, G. Polidori and R. Spagna, J. Appl. Crystallogr., 1999, 32, 115–119 CrossRef CAS.
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837–838 CrossRef CAS.
- W. H. Melhuish, J. Phys. Chem., 1961, 65, 229–235 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision B.01, Gaussian, Inc., Wallingford CT, 2010 Search PubMed.
- A. B. S. Maya, C. Perez-Melero, C. Mateo, D. Alonso, J. L. Fernandez, C. Gajate, F. Mollinedo, R. Pelaez, E. Caballero and M. Medarde, J. Med. Chem., 2005, 48, 556–568 CrossRef CAS PubMed.
- J. Y. Hu, X. Feng, A. Paudel, H. Tomiyasu, U. Rayhan, P. Thuéry, M. R. J. Elsegood, C. Redshaw and T. Yamato, Eur. J. Org. Chem., 2013, 5829–5837 CrossRef CAS.
- X. Zhou, J. He, L. S. Liao, M. Lu, X. M. Ding, X. Y. Hou, X. M. Zhang, X. Q. He and S. T. Lee, Adv. Mater., 2000, 12, 265–269 CrossRef CAS.
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877–910 CrossRef CAS PubMed.
- T. Michinobu, H. Osako and K. Shigehara, Macromolecules, 2009, 42, 8172–8180 CrossRef CAS.
- S. Janietz, D. D. C. Bradley, M. Grell, C. Giebeler, M. Inbasekaran and E. P. Woo, Appl. Phys. Lett., 1998, 73, 2453–2455 CrossRef CAS PubMed.
- J. B. Birks, Photophysics of Organic Molecules, Wiley-Interscience, London, 1970 Search PubMed.
- C. Bazzini, S. Brovelli, T. Caronna, C. Gambarotti, M. Giannone, P. Macchi, F. Meinardi, A. Mele, W. Panzeri, F. Recupero, A. Sironi and R. Tubino, Eur. J. Org. Chem., 2005, 1247–1257 CrossRef CAS.
- C. Reichardt, Solvents and Solvent Effects in Organic Chemistry, WILEY-VCH, Weinheim, 2003 Search PubMed.
- S. I. Gorelsky, SWizard program, University of Ottawa, Ottawa, Canada, 2010, http://www.sg-chem.net/ Search PubMed.
- S. I. Gorelsky and A. B. P. Lever, J. Organomet. Chem., 2001, 635, 187–196 CrossRef CAS.
- R. E. Harding, S.-C. Lo, P. L. Burn and I. D. W. Samuel, Org. Electron., 2008, 9, 377–384 CrossRef CAS PubMed.
- J. Li, P. I. Djurovich, B. D. Alleyne, M. Yousufuddin, N. N. Ho, J. C. Thomas, J. C. Peters, R. Bau and M. E. Thompson, Inorg. Chem., 2005, 44, 1713–1727 CrossRef CAS PubMed.
- C. Adachi, R. C. Kwong, P. Djurovich, V. Adamovich, M. A. Baldo, M. E. Thompson and S. R. Forrest, Appl. Phys. Lett., 2001, 79, 2082–2084 CrossRef CAS PubMed.
- Y. Z. Wu, X. Y. Zheng, W. Q. Zhu, R. G. Sun, X. Y. Jiang, Z. L. Zhang and S. H. Xu, Appl. Phys. Lett., 2003, 83, 5077–5079 CrossRef CAS PubMed.
- A. L. Fisher, K. E. Linton, K. T. Kamtekar, C. Pearson, M. R. Bryce and M. C. Petty, Chem. Mater., 2011, 23, 1640–1642 CrossRef CAS.
- Z. Zhao, X. Xu, H. Wang, P. Lu, G. Yu and Y. Liu, J. Org. Chem., 2008, 73, 594–602 CrossRef CAS PubMed.
- J. Ye, Z. Chen, M. K. Fung, C. Zheng, X. Ou, X. Zhang, Y. Yuan and C. S. Lee, Chem. Mater., 2013, 25, 2630–2637 CrossRef CAS.
- C. L. Chiang, S. M. Tseng, C. T. Chen, C. P. Hsu and C. F. Shu, Adv. Funct. Mater., 2008, 18, 248–257 CrossRef CAS.
- Y. Yang, R. C. da Costa, D. M. Smilgies, A. J. Campbell and M. J. Fuchter, Adv. Mater., 2013, 25, 2624–2628 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Mechanism of photocyclization, the photo of single-crystal for 1; 1H NMR, 13C NMR, HRMS spectra for compounds 1–5; TD-DFT calculations for 1; UV/Vis absorption and PL spectra of 2, PL quantum yields of 1 and 2. CCDC 991300. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ra13486a |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.