
Ionic liquid-based random copolymers: a new type of polymer electrolyte with low glass transition temperature
Abstract
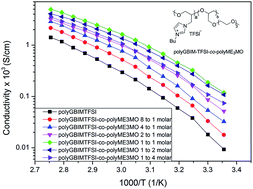
A new type of polymer electrolyte has been prepared from the side-chains of ionic liquids (IL) and an analogue of ethylene oxide (EO) directly grafted on a polyethylene oxide backbone. By tuning the two types of monomer composition during polymerization, a series of copolymers with different monomer ratios between IL : EO = 8 : 1 to 1 : 4 have been synthesized. All copolymerized polyionic liquids showed higher ionic conductivity than an IL homopolymer. By choosing a composition with IL : EO = 1 : 1, the corresponding polymer gave the highest conductivity, 1.2 × 10−4 S cm−1 at room temperature, compared to 9.3 × 10−6 S cm−1 from a homopolymer. We attribute this to one order of magnitude improvement of conductivity from a low glass transition temperature (Tg) below −40 °C.
- This article is part of the themed collection: Ionic Liquids: Editors collection for RSC Advances
 Please wait while we load your content...
Please wait while we load your content...

