DOI:
10.1039/C4RA13276A
(Paper)
RSC Adv., 2015,
5, 11534-11540
Dynamic and static behavior of the E–E′ bonds (E, E′ = S and Se) in cystine and derivatives, elucidated by AIM dual functional analysis†
Received
28th October 2014
, Accepted 5th January 2015
First published on 5th January 2015
Abstract
Atoms-in-molecules dual functional analysis (AIM-DFA) is applied to the E–E′ bonds (E, E′ = S and Se) in R-cystine (1) and the derivatives of 1, together with MeEE′Me. Hb(rc) are plotted versus Hb(rc) − Vb(rc)/2 at bond critical points (BCPs), where Hb(rc) − Vb(rc)/2 = (ħ2/8m)∇2ρb(rc). The plots are analyzed by the polar coordinate (R, θ) representation. Data of perturbed structures around the fully optimized structures are also plotted in this treatment. Perturbed structures are generated using NIV (normal coordinates of internal vibrations). Each plot for an interaction with data of a fully optimized and four perturbed structures gives a curve, which supplies important information. It is expressed by (θp, κp): θp corresponds to the tangent line for the plot measured from the y-direction and κp is the curvature. While (R, θ) correspond to the static nature of interactions, (θp, κp) represent the dynamic nature. The behavior of the E–E′ bonds is well described by (R, θ) and (θp, κp).
Introduction
The E–E′ bonds (E, E′ = S and Se) are of current and continuous interest due to their indispensable role in biological, chemical and physical sciences.1–7 The S–S bond plays a crucial role in maintaining the three dimensional structures of peptides. On the other hand, the E–E′ bonds show typical behavior in the redox process,8 which must be responsible for the highly important biological activities such as detoxification of hydroperoxides in the glutathione peroxidase (GPx) process.9–21
Scheme 1 summarizes the proposed catalytic mechanism for the antioxidant activity of GPx, which is one of the typical examples of the intervention of E–E′ in biological activities. The mechanism involves the initial oxidation of selenol (R1-SeH) to produce the corresponding selenenic acid (R1-SeOH), which reacts with glutathione (GSH) to produce selenenyl sulfide (R1-SeSG). Then a second molecule of GSH attacks at the sulfur center of R1-SeSG to regenerate the active form of the enzyme (R1-SeH) (GPx cycle in Scheme 1). In the overall process, 2 equivalent of GSH is oxidized to the corresponding disulfide (GSSG), while the hydroperoxide is reduced to water.22,23 When the peroxide concentration is higher than that of the thiol, the selenium center in the selenenic acid (R1-SeOH) may undergo further oxidation to produce the seleninic acid (R1-SeO2H) (another cycle in Scheme 1).
 |
| | Scheme 1 Proposed catalytic mechanism for the antioxidant activity of GPx. | |
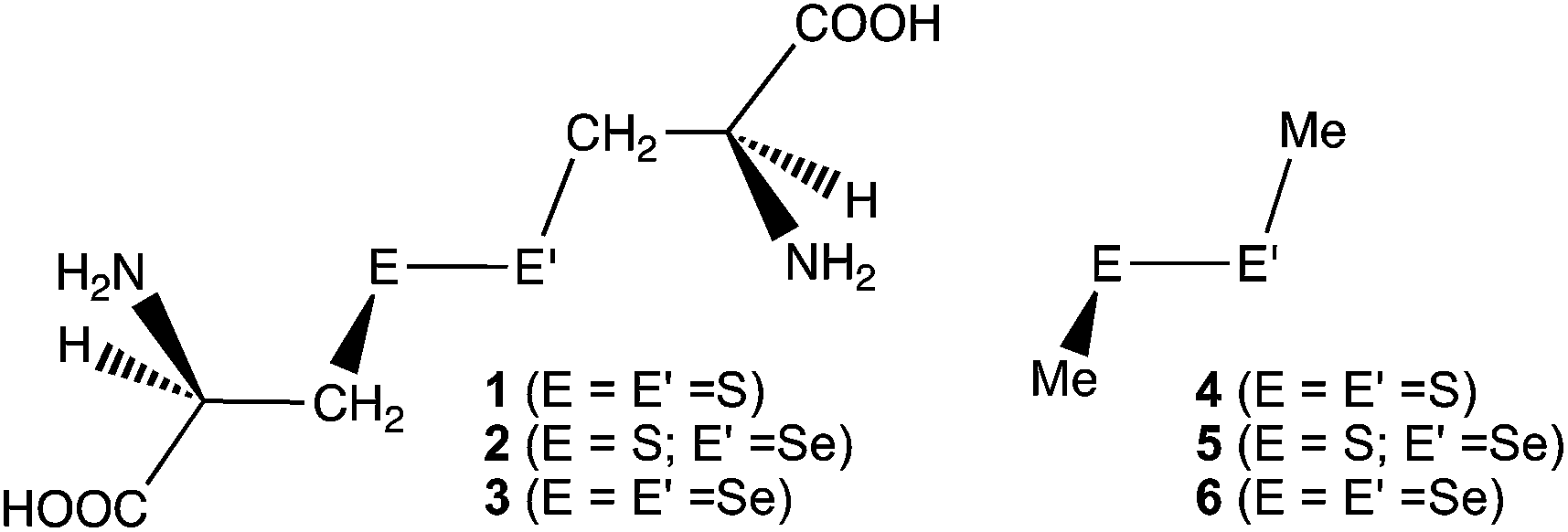
The behavior of the E–E′ bonds seems well described at first glance, however, it is still of highly importance to clarify the causality in the phenomena of the bonds, with physical necessity. Here, we clarified the dynamic and static behavior of the S–S bond in R-cystine (1), of which structures were determined by the X-ray crystallographic analysis, although the derivatives.24 Similar behavior of the S–Se and Se–Se bonds in the derivatives of 1 (2 and 3, respectively) was also investigated, together with the E–E′ bonds in MeEE′Me (E, E′ = S and Se) (4–6, respectively) (Chart 1). The effect of the hydrogen bonds (HBs) in 1–3 on the dynamic and static nature of the E–E′ bonds must also be of interest.
 |
| | Chart 1 Chalcogen–chalcogen bonds in 1–6, examined. | |
Atoms-in-molecules dual functional analysis (AIM-DFA),25–28 which we proposed recently, is applied to the E–E′ bonds in 1–6. AIM-DFA is surveyed next, together with the basic concept of the AIM approach introduced by Bader.29–31
Survey of AIM and AIM dual functional analysis
AIM (atoms-in-molecules method), proposed by Bader,29–31 enables us to analyze and evaluate the nature of chemical bonds and interactions, together with the classification.32–36 The bond critical point (BCP; rc, *) is an important concept in AIM. BCP of (ω, σ) = (3, −1)29,30 is a point along the bond path at the interatomic surface, where the electron density at BCPs (ρb(rc)) reaches a minimum. Chemical bond or interaction between A and B is denoted by A–B, which corresponds to the bond path between A and B in AIM. A notation A-*-B will be employed for A–B to emphasize the presence of BCP (*) in A–B.
Interactions are classified by signs of ∇2ρb(rc) (Laplacian ρb(rc)) and Hb(rc) (total electron energy densities). Hb(rc) are the sum of kinetic energy densities (Gb(rc)) and potential energy densities (Vb(rc)) at BCPs, as shown in eqn (1). Eqn (2) represents the relation between ∇2ρb(rc) and Hb(rc), together with Gb(rc) and/or Vb(rc), which is closely related to the Virial theorem.
| | |
Hb(rc) = Gb(rc) + Vb(rc)
| (1) |
| | |
(ħ2/8m)∇2ρb(rc) = Hb(rc) − Vb(rc)/2
| (2) |
| | |
(ħ2/8m)∇2ρb(rc) = Gb(rc) + Vb(rc)/2
| (2′) |
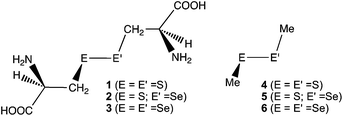
Interactions in the region of ∇2ρb(rc) < 0 are called shared-shell (SS) interactions and they are closed-shell (CS) interactions for ∇2ρb(rc) > 0. Hb(rc) must be negative when ∇2ρb(rc) < 0, since Hb(rc) are larger than (ħ2/8m)∇2ρb(rc) by Vb(rc)/2 with negative Vb(rc) at all BCPs (eqn (2)). Consequently, ∇2ρb(rc) < 0 and Hb(rc) < 0 for the SS interactions. The CS interactions are especially called pure CS interactions for Hb(rc) > 0 and ∇2ρb(rc) > 0, since electrons are destabilized and ρ(r) are depleted at (around) BCPs under the conditions.29 Electrons in the intermediate region between SS and pure CS, which belong to CS, are locally depleted but stabilized at BCPs, since ∇2ρb(rc) > 0 but Hb(rc) < 0.29 We call the interactions in this region regular CS,25–27 when it is necessary to distinguish from pure CS. The role of ∇2ρb(rc) in the classification can be replaced by Hb(rc) − Vb(rc)/2, since (ħ2/8m)∇2ρb(rc) = Hb(rc) − Vb(rc)/2 (eqn (2)).
We proposed AIM-DFA by plotting Hb(rc) versus Hb(rc) − Vb(rc)/2.26,27 Both axes in the plot are given in energy unit, therefore, distances on the (x, y) [= (Hb(rc) − Vb(rc)/2, Hb(rc))] plane can be expressed in the energy unit, which provides an analytical development. AIM-DFA incorporates the classification of interactions by signs of ∇2ρb(rc) [= (8m/ħ2)(Hb(rc) − Vb(rc)/2)] and Hb(rc). Scheme 2 summarizes the treatment. Interactions of pure CS appear in the first quadrant, those of regular CS in the forth quadrant and data of the SS interactions drop in the third quadrant. No interactions appear in the second one.
| | |
θp = 90° − tan−1(dy/dx)
| (5) |
| | |
κp = |d2y/dx2|/[1 + (dy/dx)2]3/2
| (6) |
where (
x,
y) = (
Hb(
rc) −
Vb(
rc)/2,
Hb(
rc)) and
y/
x =
Hb(
rc)/(
Hb(
rc) −
Vb(
rc)/2).
 |
| | Scheme 2 AIM-DFA treatment of interactions: plot of Hb(rc) versus Hb(rc) − Vb(rc)/2 for weak to strong interactions. | |
In our treatment, data for perturbed structures around fully optimized ones are also employed for the plots, together with the fully optimized structures.25–28 The method to generate the perturbed structures will be discussed later. The plots of Hb(rc) versus Hb(rc) − Vb(rc)/2 are analyzed employing the polar coordinate (R, θ) representation with the (θp, κp) parameters.26–28 The treatment is explained in Fig. 1. R in (R, θ) is defined by eqn (3) and given in the energy unit. R corresponds to the energy for an interaction at BCP. The plots for weak to strong interactions show a spiral stream, as a whole. θ in (R, θ) is defined by eqn (4) and measured from the y-axis. θ controls the spiral stream of the whole plots. Each plot for an interaction shows a specific curve, which provides important information.26,27,37 The curve is expressed by (θp, κp). θp is defined by eqn (5) and measured from the y-direction, which corresponds to the tangent line of the plot. κp is the curvature of the plot (eqn (6)).28 While (R, θ) correspond to the static nature of interactions, (θp, κp) represent the dynamic nature. Indeed, (θp, κp) originate mainly from the data of perturbed structures, but they are recognized as the nature at the fully optimized structure. k defined by eqn (7) is also an important and useful parameter in AIM analysis. AIM-DFA will provide an excellent possibility to evaluate, understand and classify weak to strong interactions in a unified form.
 |
| | Fig. 1 Polar (R, θ) coordinate representation of Hb(rc) versus Hb(rc) − Vb(rc)/2, with the (θp, κp) parameters. | |
Methodological details of QC and AIM calculations
Structures of 1–6 were optimized using the Gaussian 09 program package,38 after the conformation research with the Monte-Carlo method39 in the Spartan 02 (ref. 40) for 1–3. At least seven hundred and twenty two conformers were generated for each with the PM3 method.41 Thirty of most stable conformers by the Monte-Carlo method were re-optimized using the 6-311+G(3d) basis sets42 for S and Se and the 6-311++G(d,p) basis sets for O, N, C and H at the M062X level.43 Frequency analysis was performed on three of most stable conformers at the same method for each of 1a–3c, where a represents the most stable conformer with the second and third ones by b and c, respectively. The global minima for 4–6 were determined by usual optimizations with the frequency analysis.
AIM parameters were calculated with the Gaussian 09 program package and analyzed by the AIM2000 program.44 Normal coordinates of the internal vibrations (NIV) are employed to generate the perturbed structures necessary to evaluate the dynamic nature of the interactions.28 The k-th perturbed structures in question (Skw) will be given by the addition of the k-th NIV (Nk) to the coordinates of the standard orientation at the fully optimized structure (So) in the matrix representation.28 Eqn (8) explains the treatment. The r(E, E′) values in the perturbed structures were determined as shown by eqn (9) or (9′), where ao is the Bohr radius (0.52918 Å) with to (E, E′) in the fully optimized structure. Data at w = 0, ±0.05 and ±0.1 were employed for the analysis.26,27,45 The k-th internal vibration (νn) must be located most effectively on E–E′, which corresponding to the selected Nk.
| | |
r(E, E′) = ro(E, E′) + wao
| (9) |
Results and discussion
Fig. 2 draws the three of most stable conformers for 1–3 (1a–3c). Fig. 2 also shows the stretching modes of 1a–3c, necessary to evaluate the dynamic nature of the E–E′ bonds with NIV. Conformers 1a–3c are stabilized by intramolecular HBs, as shown in Fig. 2. Table 1 collects the optimized E–E′ distances and torsional angles (ϕ(CEE′C)) for 1–6, together with the distances and angles for the intramolecular HBs formed between the different moieties of the E–E′ bonds in 1a–3c.
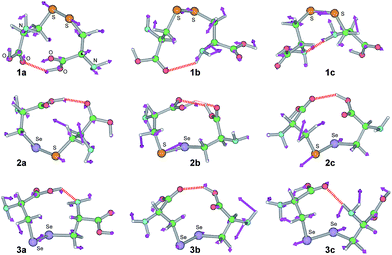 |
| | Fig. 2 Optimized structures for 1a–3c. Directions of the motions, corresponding to NIV used to generate the perturbed structures, are shown (purple arrows), together with the intramolecular hydrogen bonds (red heavy dotted lines). | |
Table 1 Optimized E–E′ distances and torsional angles for 1–6, together with the distances and angles for the intramolecular hydrogen bonds in 1a–3ca
| Speciesb |
ΔErel (kJ mol−1) |
ro(E, E′) (Å) |
ϕ(CEE′C) (°) |
(A–H⋯B) |
ro(A, H) (Å) |
ro(B, H) (Å) |
∠AHB (°) |
| The 6-311+G(3d) basis sets being employed for S and Se with the 6-311++G(d,p) basis sets for O, N, C and H at the DFT level of M062X. RSH = R-cysteine, for example. |
| RS-*-SR (1a: C1) |
0.0 |
2.0625 |
67.7 |
(O–H⋯O) |
0.9780 |
1.9828 |
149.8 |
| RS-*-SR (1b: C1) |
0.3 |
2.0471 |
−82.2 |
(N–H⋯O) |
1.0155 |
2.4136 |
134.4 |
| RS-*-SR (1c: C1) |
0.7 |
2.0529 |
88.5 |
(N–H⋯N) |
1.0210 |
2.2551 |
172.3 |
| RS-*-SeR (2a: C1) |
0.0 |
2.1984 |
−83.9 |
(O–H⋯O) |
0.9850 |
1.8025 |
156.4 |
| RS-*-SeR (2b: C1) |
15.7 |
2.1890 |
84.3 |
(O–H⋯O) |
0.9748 |
1.8996 |
142.6 |
| |
|
|
|
(O–H⋯O) |
0.9743 |
2.1631 |
126.4 |
| RS-*-SeR (2c: C1) |
17.5 |
2.2011 |
94.0 |
(O–H⋯O) |
0.9771 |
1.8581 |
142.9 |
| RSe-*-SeR (3a: C1) |
0.0 |
2.3275 |
88.5 |
(O–H⋯N) |
0.9863 |
1.8955 |
137.7 |
| RSe-*-SeR (3b: C1) |
1.4 |
2.3303 |
93.4 |
(O–H⋯O) |
0.9773 |
1.8335 |
145.1 |
| RSe-*-SeR (3c: C1) |
3.3 |
2.3309 |
90.2 |
(N–H⋯O) |
1.0193 |
2.0185 |
167.6 |
| MeS-*-SMe (4: C2) |
|
2.0491 |
85.0 |
|
|
|
|
| MeSe-*-SMe (5: C1) |
|
2.1923 |
85.6 |
|
|
|
|
| MeSe-*-SeMe (6: C2) |
|
2.3236 |
86.1 |
|
|
|
|
Fig. 3 shows contour maps of ρb(rc) drawn on an EE′C plane of 1a–3c. All BCPs expected are clearly detected, containing those on the E–E′, E–C (E′–C) and C–H bonds, together with HBs. AIM-DFA is applied to clarify the static and dynamic behavior of E–E′ in 1–6. Fig. 4 shows the plots of Hb(rc) versus Hb(rc) − Vb(rc)/2 for 1–6. The magnified picture around the data of the fully optimized structures for 2a–2c and 5 is also shown in Fig. 4. AIM parameters corresponding to the static and dynamic behavior of E–E′ are obtained for 1–6, through analysis of the plots in Fig. 4, according to eqn (3)–(6). Table 2 collects the AIM functions and parameters of the static behavior evaluated for 1–6. Table 3 summarizes the AIM parameters of the dynamic behavior evaluated for 1–6.
 |
| | Fig. 3 Contour maps of ρb(rc) drawn on the E–E′–C planes of 1a–3c, together with BCPs (red solid circles), RCPs (ring critical points: lime green solid squares), and bond paths (solid lines). The counters (eao−3) are at 2l (l = ±8, ±7, …, 0) and 0.0047, which corresponds to the molecular surface (heavy line). | |
 |
| | Fig. 4 Plots of Hb(rc) versus Hb(rc) − Vb(rc)/2 for 1–6. (a) Whole picture and (b) magnified one around data of fully optimized structures for 2a–2c and 5. | |
Table 2 Static nature of the E–E′ bonds in 1–6 at full optimized structuresa
| Speciesb |
ρb(rc) (au) |
c∇2ρb(rc)c (au) |
Hb(rc) (au) |
Rd (au) |
θe (°) |
kf |
| The 6-311+G(3d) basis sets being employed for S and Se with the 6-311++G(d,p) basis sets for O, N, C and H at the DFT level of M062X. RSH = R-cysteine, for example. c∇2ρb(rc) = Hb(rc) − Vb(rc)/2 where c = ħ2/8m. R = [(Hb(rc) − Vb(rc)/2)2 + Hb(rc)2]1/2 (eqn (3)). θ = 90° − tan−1[Hb(rc)/(Hb(rc) − Vb(rc)/2)] (eqn (4)). k = Vb(rc)/Gb(rc) (eqn (7)). |
| RS-*-SR (1a: C1) |
0.1409 |
−0.0117 |
−0.0710 |
0.0719 |
189.4 |
−2.495 |
| RS-*-SR (1b: C1) |
0.1443 |
−0.0126 |
−0.0749 |
0.0759 |
189.6 |
−2.509 |
| RS-*-SR (1c: C1) |
0.1432 |
−0.0124 |
−0.0737 |
0.0747 |
189.6 |
−2.508 |
| RS-*-SeR (2a: C1) |
0.1171 |
−0.0041 |
−0.0529 |
0.0531 |
184.4 |
−2.183 |
| RS-*-SeR (2b: C1) |
0.1188 |
−0.0045 |
−0.0547 |
0.0548 |
184.7 |
−2.195 |
| RS-*-SeR (2c: C1) |
0.1166 |
−0.0040 |
−0.0525 |
0.0527 |
184.4 |
−2.180 |
| RSe-*-SeR (3a: C1) |
0.1020 |
−0.0042 |
−0.0431 |
0.0433 |
185.6 |
−2.242 |
| RSe-*-SeR (3b: C1) |
0.1021 |
−0.0046 |
−0.0432 |
0.0434 |
186.0 |
−2.268 |
| RSe-*-SeR (3c: C1) |
0.1023 |
−0.0046 |
−0.0435 |
0.0437 |
186.1 |
−2.269 |
| MeS-*-SMe (4: C2) |
0.1446 |
−0.0131 |
−0.0751 |
0.0763 |
189.9 |
−2.535 |
| MeSe-*-SMe (5: C1) |
0.1189 |
−0.0048 |
−0.0544 |
0.0547 |
185.0 |
−2.213 |
| MeSe-*-SeMe (6: C2) |
0.1036 |
−0.0050 |
−0.0445 |
0.0448 |
186.4 |
−2.291 |
Table 3 Dynamic nature of the E–E′ bonds in 1–6 with NIVa
| Speciesb |
νnc (cm−1) |
n for νn |
kfd (mdyn Å−1) |
θpe (°) |
κpf (au−1) |
| The 6-311+G(3d) basis sets being employed for S and Se with the 6-311++G(d,p) basis sets for O, N, C and H at the DFT level of M062X. RSH = R-cysteine, for example. Frequency corresponding to the stretching mode of the E-*-E′ bond, where * means the bond critical point in question. Force constants correspond to νn. θp = 90° − tan−1(dy/dx) where (x, y) = (Hb(rc) − Vb(rc)/2, Hb(rc)) (eqn (5)). kp = |d2y/dx2|/[1 + (dy/dx)2]3/2 (eqn (6)). |
| RS-*-SR (1a: C1) |
511.3 |
20 |
2.060 |
197.5 |
0.75 |
| RS-*-SR (1b: C1) |
522.5 |
21 |
1.118 |
197.4 |
0.67 |
| RS-*-SR (1c: C1) |
506.8 |
20 |
1.947 |
197.5 |
0.69 |
| RS-*-SeR (2a: C1) |
414.4 |
18 |
0.551 |
188.0 |
0.34 |
| RS-*-SeR (2b: C1) |
423.6 |
19 |
1.896 |
188.4 |
0.33 |
| RS-*-SeR (2c: C1) |
414.7 |
19 |
1.440 |
188.2 |
0.40 |
| RSe-*-SeR (3a: C1) |
301.9 |
15 |
1.875 |
188.9 |
0.71 |
| RSe-*-SeR (3b: C1) |
308.0 |
16 |
0.174 |
189.3 |
0.77 |
| RSe-*-SeR (3c: C1) |
298.7 |
13 |
0.938 |
189.4 |
0.68 |
| MeS-*-SMe (4: C2) |
513.7 |
6 |
2.645 |
197.6 |
0.66 |
| MeS-*-SeMe (5: C1) |
419.7 |
6 |
2.072 |
188.6 |
0.38 |
| MeSe-*-SeMe (6: C2) |
307.7 |
6 |
2.730 |
189.1 |
0.77 |
The behavior of E–E′ in 1–6 is examined by comparing the (θ, θp, R) values with those of the standard ones. The standard values are roughly determined to classify the interactions in question, employing typical weak to strong interactions. Scheme 3 illustrates the standard values for the typical interactions. The results tell us that θ are larger than 180° for SS interactions, which correspond to Hb(rc) − Vb(rc)/2 < 0. The θp value will play an important role to discuss the characters of CS interactions with θ < 180° (Hb(rc) − Vb(rc)/2 > 0). While the character of interactions will be that of CT-TBP (trigonal bipyramidal adducts formed through charge-transfer) such as in Me2Se(-*-Cl)2, if θp > 180°, it will be that of CT-MC (molecular complexes formed through CT) such as in Me2S-*-Br2, when θp < 180°, for example. The R values contribute to classify SS, further. Classical chemical bonds of SS are strong for R > 0.15 au but they would be weak when R < 0.15 au.26
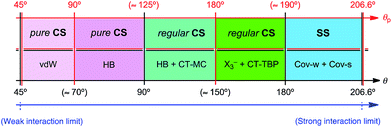 |
| | Scheme 3 Rough classification of interactions by θ and θp. | |
The (θ/°, θp/°, R/au) values for S–S in 1a and 4 are (189.4, 197.5, 0.072) and (189.9, 197.6, 0.076), respectively. While the values for S–Se in 2a and 5 are (184.4, 188.0, 0.053) and (185.0, 188.6, 0.055), respectively, those for Se–Se in 3a and 6 are (185.6, 188.9, 0.043) and (186.4, 189.1, 0.045), respectively. The results show that all E–E′ in 1–6 are classified as the weak covalent bonds, since θ > 180° with R < 0.15 au.26 The nature of E–E′ in 1a–3a is predicted to be very close to that of 4–6, respectively. The strength of E–E′ is reconfirmed in the order of Se–Se ≤ S–Se < S–S. All data of Se–Se, S–Se and S–S in 1–6 appear in the SS region. However, those of Se–Se and S–Se are closer to the regular CS region (θp ≈ 189°), relative to the case of S–S (θp ≈ 198°). Whereas R for 1a–3a are less than those of 4–6, respectively, the data for 1a–3a seem to appear at the opposite side of the origin in the plots. The discrepancies must be the reflection of the complex contributions from Gb(rc) and Vb(rc) to Hb(rc) and Hb(rc) − Vb(rc)/2 in E–E′, according to eqn (1) and (2). It would be difficult to specify the reason for the characteristic behaviour in the plots, based on the data in Tables 2 and 3.
How are the E–E′ bonds affected by the formation of HBs? The formation of intramolecular HBs stabilizes 1–3, relative to the conformers with no such interactions. The A–H⋯B angles are predicted to be around 130–150° for HBs in 1–3, which must be the reflection of the restricted HBs in 1–3. The results may suggest that the intramolecular HBs are formed under somewhat undesirable conditions (see Table 1). The HBs in 1–3 must affect on the stability and the strength of E–E′ in 1–3. The variation of ϕ(CEE′C) in 1–3, relative to the case of 4–6, must be another evidence for the restricted HBs (Table 1). The conformation also affect on the strength of E–E′. In the case of 2, the S–Se bond becomes stronger in the order of 2c ≤ 2a < 2b, although very slightly (see Fig. 4b, Tables 2 and 3). The strength of S–Se seems almost independent of the stability in 2a–c, for example. Namely, such molecules are stabilized through the formation of HBs, but the E–E′ bonds could be sacrificed and somewhat weakened by the distortion.
The nature of the E–E′ bonds in 1–6 is well described with the dynamic nature of (θp, κp) and the static nature of (R, θ) by applying AIM-DFA.
Conclusion
AIM-DFA is applied to the E–E′ bonds (E, E′ = S and Se) in R-cystine (1) and the derivatives (2 and 3 for the S–Se and Se–Se derivatives, respectively), together with those of MeEE′Me (4–6). The dynamic and static behavior is clarified for E–E′ in 1–6 by the application. The nature of E–E′ in 1–6 is further examined by comparing the (θ, θp, R) values with those of the standard ones. The (θ/°, θp/°, R/au) values for S–S in 1a and 4 are (189.4, 197.5, 0.072) and (189.9, 197.6, 0.076), respectively. While the values for S–Se in 2a and 5 are (184.4, 188.0, 0.053) and (185.0, 188.6, 0.055), respectively, those for Se–Se in 3a and 6 are (185.6, 188.9, 0.043) and (186.4, 189.1, 0.045), respectively. All E–E′ in 1–6 are classified as the weak covalent bonds. The nature of E–E′ in 1a–3a is very close to that in 4–6, respectively. The strength of the bonds is predicted to be in the order of S–S > S–Se ≥ Se–Se. All data of E–E′ in 1–6 appear in the SS region. However, those of Se–Se and S–Se are closer to the regular CS region (θp ≈ 189°), relative to the case of S–S (θp ≈ 198°). The A–H⋯B angles in HBs of 1–3 are predicted to be around 130–150°, which must be the reflection of the restricted HBs in 1–3. Indeed, 1–3 are stabilized by the formation of HBs, but the E–E′ bonds could be sacrificed and somewhat weakened by the distortion. The nature of the E–E′ bonds in 1–6 is well described by (θp, κp) of the dynamic nature and (R, θ) of the static nature obtained through AIM-DFA.
Acknowledgements
This work was partially supported by a Grant-in-Aid for Scientific Research (nos 23350019 and 26410050) from the Ministry of Education, Culture, Sports, Science and Technology, Japan. The support of the Wakayama University Original Research Support Project Grant and the Wakayama University Graduate School Project Research Grant is also acknowledged.
Notes and references
-
(a) Organic Selenium Compounds: Their Chemistry and Biology, ed. D. L. Klayman and W. H. H. Günther, Wiley, New York, 1973 Search PubMed;
(b) The Chemistry of Organic Selenium and Tellurium Compounds, ed. S. Patai and Z. Rappoport, John-Wiley and Sons, New York, 1986, vol. 1 and 2 Search PubMed;
(c) Organic Selenium Chemistry, ed. D. Liotta, Wiley-Interscience, New York, 1987 Search PubMed;
(d) Organoselenium Chemistry, A practical Approach, ed. T. G. Back, Oxford University Press, Oxford, 1999 Search PubMed;
(e) Organoselenium Chemistry Modern Developments in Organic Synthesis, Topics in Current Chemistry, ed. T. Wirth, Springer, Berlin, Heidelberg, New York, London, Paris, Tokyo, 2000 Search PubMed.
- Chemistry of Hypervalent Compounds, ed. K.-y. Akiba, Wiley-VCH, New York, 1999 Search PubMed.
-
(a) W. Nakanishi, Hypervalent Chalcogen Compounds, in Handbook of Chalcogen Chemistry: New Perspectives in Sulfur, Selenium and Tellurium, ed. F. A. Devillanova, Royal Society of Chemistry, Cambridge, 2006, ch. 10.3, pp. 644–668 Search PubMed;
(b) W. Nakanishi and S. Hayashi, Hypervalent Chalcogen Compounds, in Handbook of Chalcogen Chemistry: New Perspectives in Sulfur, Selenium and Tellurium, ed. F. A. Devillanova and W.-W. du Mont, Royal Society of Chemistry, Cambridge, 2nd edn, 2013, vol. 2, ch. 12.3, pp. 335–372 Search PubMed.
- A. J. Mukherjee, S. S. Zade, H. B. Singh and R. B. Sunoj, Chem. Rev., 2010, 110, 4357 CrossRef CAS PubMed.
- M. Kulcsar, A. Beleaga, C. Silvestru, A. Nicolescu, C. Deleanu, C. Todasca and A. Silvestru, Dalton Trans., 2007, 2187 RSC; A. Beleaga, M. Kulcsar, C. Deleanu, A. Nicolescu, C. Silvestru and A. Silvestru, J. Organomet. Chem., 2009, 694, 1308 CrossRef CAS.
- S. Hayashi and W. Nakanishi, J. Org. Chem., 1999, 64, 6688 CrossRef CAS PubMed; W. Nakanishi, S. Hayashi and S. Toyota, J. Org. Chem., 1998, 63, 8790 CrossRef; W. Nakanishi, S. Hayashi and S. Toyota, Chem. Commun., 1996, 371 RSC; W. Nakanishi, S. Hayashi and H. Yamaguchi, Chem. Lett., 1996, 947 CrossRef; W. Nakanishi, Chem. Lett., 1993, 2121 CrossRef.
- W. Nakanishi, S. Hayashi and T. Arai, Chem. Commun., 2002, 2416 RSC.
- W. Nakanishi, S. Hayashi, S. Morinaka, T. Sasamori and N. Tokitoh, New J. Chem., 2008, 32, 1881 RSC.
-
(a) W. Brandt and L. A. Wessjohann, ChemBioChem., 2005, 6, 386 CrossRef CAS PubMed;
(b) S. Gromer, L. A. Wessjohann, J. Eubel and W. Brandt, ChemBioChem., 2006, 7, 1649 CrossRef CAS PubMed;
(c) L. A. Wessjohann, A. Schneider, M. Abbas and W. Brandt, Biol. Chem., 2007, 388, 997 CrossRef CAS PubMed;
(d) B. M. Lacey, B. E. Eckenroth, S. Flemer Jr and R. J. Hondal, Biochemistry, 2008, 47, 12810 CrossRef CAS PubMed;
(e) L. A. Wessjohann and A. Schneider, Chem. Biodiversity, 2008, 5, 375 CrossRef CAS PubMed.
- B. Ren, W. Huang, B. Åkesson and R. Ladenstein, J. Mol. Biol., 1997, 268, 869 CrossRef CAS PubMed.
- W. A. Hendrickson, Science, 1991, 254, 51 CAS.
- A. Ishii, S. Matsubayashi, T. Takahashi and J. Nakayama, J. Org. Chem., 1999, 64, 1084 CrossRef CAS.
- T. Saiki, K. Goto and R. Okazaki, Angew. Chem., Int. Ed., 1997, 36, 2223 CrossRef CAS; K. Goto, D. Sonoda, K. Shimada, S. Sase and T. Kawashima, Angew. Chem., Int. Ed., 2010, 49, 545 CrossRef PubMed.
- G. Mugesh and H. B. Singh, Chem. Soc. Rev., 2000, 29, 347 RSC.
- K. P. Bhabak and G. Mugesh, Chem.–Asian J., 2009, 4, 974 CrossRef CAS PubMed; B. K. Sarma and G. Mugesh, Chem.–Eur. J., 2008, 14, 10603 CrossRef PubMed.
- K. P. Bhabak and G. Mugesh, Chem.–Eur. J., 2009, 15, 9846 CrossRef CAS PubMed; K. P. Bhabak and G. Mugesh, Chem.–Eur. J., 2008, 14, 8640 CrossRef PubMed; K. P. Bhabak and G. Mugesh, Chem.–Eur. J., 2007, 13, 4594 CrossRef PubMed; B. K. Sarma and G. Mugesh, Inorg. Chem., 2006, 45, 5307 CrossRef PubMed; P. P. Phadnis and G. Mugesh, Org. Biomol. Chem., 2005, 3, 2476 Search PubMed.
- M. Iwaoka, R. Ooka, T. Nakazato, S. Yoshida and S. Oishi, Chem. Biodiversity, 2008, 5, 359 CAS.
- F. Kumakura, B. Mishra, K. I. Priyadarsini and M. Iwaoka, Eur. J. Org. Chem., 2010, 440 CrossRef CAS.
- K. Arai, K. Dedachi and M. Iwaoka, Chem.–Eur. J., 2011, 17, 481 CrossRef CAS PubMed.
- S. Yoshida, F. Kumakura, I. Komatsu, K. Arai, Y. Onuma, H. Hojo, B. G. Singh, K. I. Priyadarsini and M. Iwaoka, Angew. Chem., Int. Ed., 2011, 50, 2125 (Angew. Chem., 2011, 123, 2173) CrossRef CAS PubMed.
- D. Manna and G. Mugesh, J. Am. Chem. Soc., 2012, 134, 4269 CrossRef CAS PubMed.
- L. Flohé, E. A. Günzler and H. H. Schock, FEBS Lett., 1973, 32, 132 CrossRef.
- L. Flohé, Glutathione peroxidase brought into focus, in Free radicals in biology, ed. W. A. Pryor, Academic Press, New York, 1982, vol. 5, pp. 223–253 Search PubMed.
- V. S. Minkov and E. V. Boldyreva, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2009, 65, o245 CAS; T. N. Drebushchak, S. V. Bizyaev and E. V. Boldyreva, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2008, 64, o313 Search PubMed; V. S. Minkov and E. V. Boldyreva, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2008, 64, o344 Search PubMed.
- W. Nakanishi, S. Hayashi and K. Narahara, J. Phys. Chem. A, 2008, 112, 13593 CrossRef CAS PubMed.
- W. Nakanishi, S. Hayashi and K. Narahara, J. Phys. Chem. A, 2009, 113, 10050 CrossRef CAS PubMed.
- W. Nakanishi and S. Hayashi, Curr. Org. Chem., 2010, 14, 181 CrossRef CAS.
- W. Nakanishi and S. Hayashi, J. Phys. Chem. A, 2010, 114, 7423 CrossRef CAS PubMed.
- Atoms in Molecules. A Quantum Theory, ed. R. F. W. Bader, Oxford University Press, Oxford, 1990 Search PubMed.
- The Quantum Theory of Atoms in Molecules: From Solid State to DNA and Drug Design, ed. C. F. Matta and R. J. Boyd, Wily-VCH, Weinheim, Germany, 2007, ch. 1 Search PubMed.
-
(a) F. Biegler-König and J. Schönbohm, J. Comput. Chem., 2002, 23, 1489 CrossRef PubMed;
(b) F. Biegler-König, J. Schönbohm and D. Bayles, J. Comput. Chem., 2001, 22, 545 CrossRef;
(c) R. F. W. Bader, J. Phys. Chem. A, 1998, 102, 7314 CrossRef CAS;
(d) R. F. W. Bader, Chem. Rev., 1991, 91, 893 CrossRef CAS;
(e) R. F. W. Bader, Acc. Chem. Res., 1985, 18, 9 CrossRef CAS;
(f) T. H. Tang, R. F. W. Bader and P. MacDougall, Inorg. Chem., 1985, 24, 2047 CrossRef CAS;
(g) R. F. W. Bader, T. S. Slee, D. Cremer and E. Kraka, J. Am. Chem. Soc., 1983, 105, 5061 CrossRef CAS;
(h) F. Biegler-König, R. F. W. Bader and T. H. Tang, J. Comput. Chem., 1982, 3, 317 CrossRef.
- J. M. Molina and J. A. Dobado, Theor. Chem. Acc., 2001, 105, 328 CrossRef CAS.
- J. A. Dobado, H. Martînez-Garcîa, J. M. Molina and M. R. Sundberg, J. Am. Chem. Soc., 2000, 122, 1144 CrossRef CAS.
- S. K. Ignatov, N. H. Rees, B. R. Tyrrell, S. R. Dubberley, A. G. Razuvaev, P. Mountford and G. I. Nikonov, Chem.–Eur. J., 2004, 10, 4991 CrossRef CAS PubMed.
- S. K. Tripathi, U. Patel, D. Roy, R. B. Sunoj, H. B. Singh, G. Wolmershäuser and R. J. Butcher, J. Org. Chem., 2005, 70, 9237 CrossRef CAS PubMed.
- R. J. Boyd and S. C. Choi, Chem. Phys. Lett., 1986, 129, 62 CrossRef CAS.
- See Fig. 2 in W. Nakanishi and S. Hayashi, J. Phys. Chem. A, 2013, 117, 1795 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- R. W. B. Ardill, K. J. M. Moriarty and M. Creutz, Comput. Phys. Commun., 1983, 29, 97 CrossRef CAS; M. Creutz, Phys. Rev. Lett., 1983, 50, 1411 CrossRef , see also P. Bratley, B. L. Fox and L. E. Schrage, A Guide to Simulation, Springer-Verlag, New York, 1987 Search PubMed; G. M. Kalos and P. A. Whitlock, Monte Carlo Methods, John Wiley & Sons, New York, 1986 Search PubMed; A. R. Leach, Molecular Modelling. Principles and Applications, Addison Wesley Longman, Essex, England, 1996 Search PubMed.
- Spartan ’02 for Windows, Wavefunction, Inc., Irvine, CA, Spartan '02 Windows, Tutorial and User's Guide, Wavefunction, Inc., Irvine, CA, 2001.
- J. J. P. Stewart, J. Comput. Chem., 1989, 10, 209 CrossRef CAS.
- In our experience, the 6-311+G(d) basis sets or higher ones are recommended, when usual organic selenium compounds are calculated. For the 6-311+G(3d) basis sets, see: R. C. Binning Jr and L. A. Curtiss, J. Comput. Chem., 1990, 11, 1206 CrossRef CAS; L. A. Curtiss, M. P. McGrath, J.-P. Blaudeau, N. E. Davis, R. C. Binning Jr and L. Radom, J. Chem. Phys., 1995, 103, 6104 CrossRef; M. P. McGrath and L. Radom, J. Chem. Phys., 1991, 94, 511 CrossRef , for the diffuse functions (+ and ++), see T. Clark, J. Chandrasekhar, G. W. Spitznagel and P. v. R. Schleyer, J. Comput. Chem., 1983, 4, 294 CrossRef , see for example W. Nakanishi and S. Hayashi, J. Phys. Chem. A, 1999, 103, 6074 CrossRef.
- Y. Zhao and G. D. Truhlar, Org. Lett., 2006, 8, 5753 CrossRef CAS PubMed; E. S. Wheeler and N. K. Houk, J. Chem. Theory Comput., 2010, 6, 395 CrossRef PubMed; S. Rayne and K. Forest, J. Mol. Struct.: THEOCHEM, 2010, 948, 102 CrossRef; S. Rayne and K. Forest, Nat. Prec., 2010 DOI:10.1038/npre.2010.4865.1.
- The AIM2000 program (Version 2.0) is employed to analyze and visualize atoms in molecules: F. Biegler-König, J. Comput. Chem., 2000, 21, 1040 CrossRef , see also ref. 31b.
- The bond orders become 1.50 and 0.67 times larger than the initial value if they are calculated at 0.2ao shorter and longer distances from the initial length, respectively.46 The change in the angles could be observed and seems to affect not so much on our discussion to classify various interactions.28,47.
- L. Pauling The Nature of the Chemical Bond, Cornell University Press, Ithaca, New York, 3rd edn, 1960, ch. 7, pp. 221–264 CrossRef CAS; L. Pauling, J. Am. Chem. Soc., 1947, 69, 542 CrossRef CAS.
- See also, W. Nakanishi, S. Hayashi, K. Matsuiwa and M. Kitamoto, Bull. Chem. Soc. Jpn., 2012, 85, 1293 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The fully optimized structures given by Cartesian coordinates for examined molecules and adducts. See DOI: 10.1039/c4ra13276a |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.