DOI:
10.1039/C4RA13236J
(Paper)
RSC Adv., 2015,
5, 477-484
Comparative study of ring-opening polymerization of L-lactide and ε-caprolactone using zirconium hexadentate bis(aminophenolate) complexes as catalysts†
Received
27th October 2014
, Accepted 20th November 2014
First published on 24th November 2014
Abstract
A series of zirconium bis(aminophenolate) complexes as catalysts for the ring opening polymerization of L-lactide (LA) and ε-caprolactone (CL) were investigated. Ligands bearing various chelating groups have a profound influence on the catalysis results. Among them, the thiophen-2-yl methyl group showed the greatest activity while the pyridine-2-yl methyl group showed the worst performance with regard to the rate of CL polymerization. However, the trend was reversed for the rate of LA polymerization. The kinetic results indicated a first-order dependency on [CL] and [LA]. However, the order of the catalyst concentration was different. Polymerization proceeded with second-order dependence on [LOMeZr(OBn)2] for CL but with first-order dependence on [LOMeZr(OBn)2] for LA.
Introduction
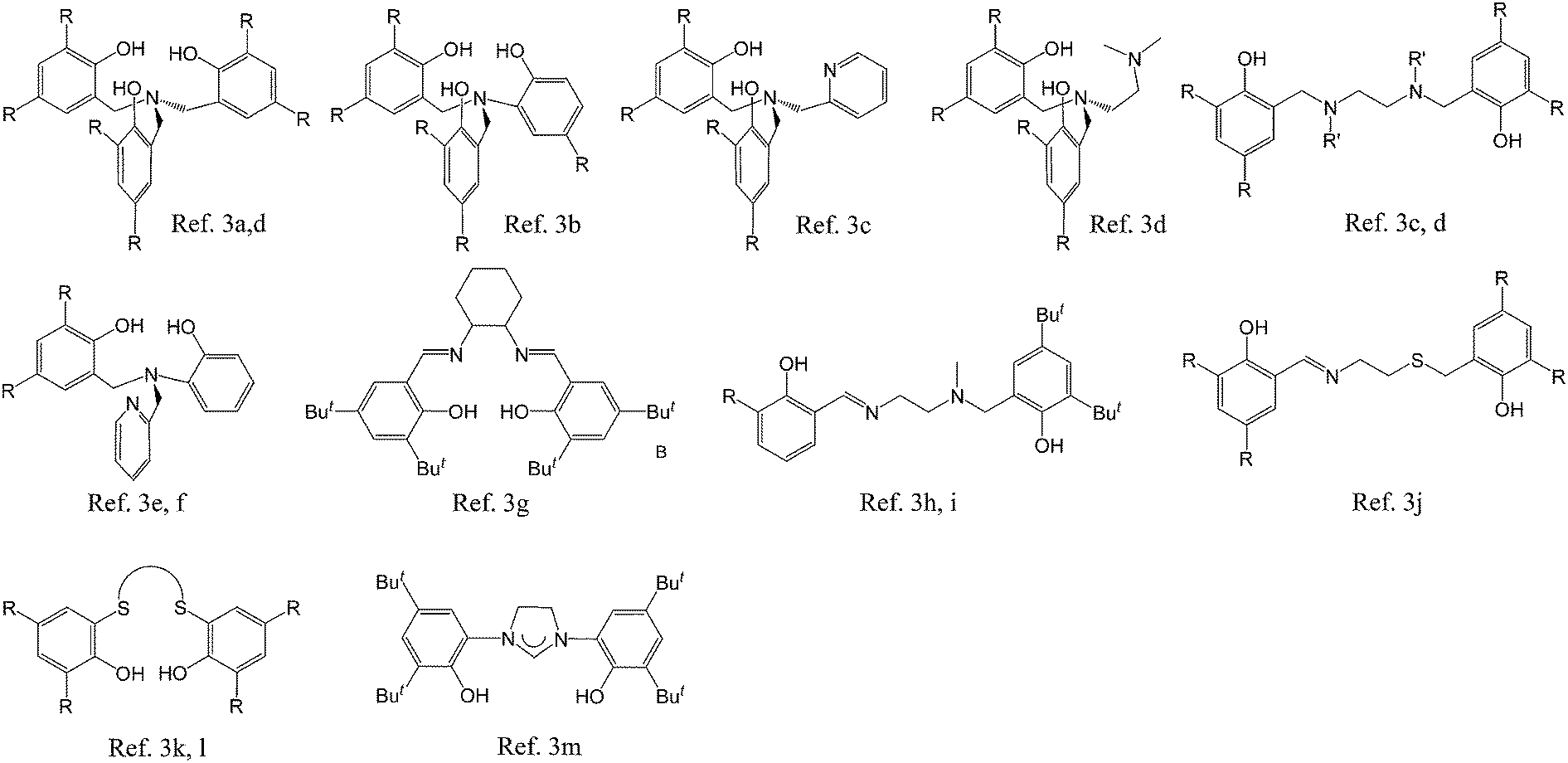
Poly(lactide) (PLA), poly(ε-caprolactone) (PCL), and their copolymers are applied in a wide range of fields1 due to their biodegradability, biocompatibility, and permeability. The most common method used for the synthesis of PLA and PCL is ring-opening polymerization (ROP). Most metal complexes2 have been used as catalysts for the ROP of cycloesters. However, the use of catalysts of low cytotoxicity is essential for materials with medicinal applications. Zirconium complexes are also commonly used as catalysts for ROP due to the inexpensive precursors and high oxidation state associated with polyanionic ligands. Fig. 1 presents a series of multi-dentate ligands of zirconium complexes used for ROP of cycloesters.
 |
| | Fig. 1 Multi-dentate ligands applied to the synthesis of zirconium complexes. | |
Among these, salen,3g salan3c,d and salalen3h,i type (i.e. bis(iminophenol)) ligands are the most commonly used due to the ease of diverse modification on the moiety between the two nitrogen atoms.4 The previous studies usually involved up to three or four dentate ligands around the metal. Never has hexadentate ligand been applied to the synthesis of zirconium complex.
In 2010 and 2012, Sun3e and Okuda3k reported zirconium complexes with an eight-coordinate metal center that included two tetradentate ligands and two OSSO bis(phenolate) ligands, respectively, which demonstrated outstanding polymerization activity for meso-lactide. These findings inspired us to design a series LZr(OR)2 complexes bearing hexadentate salan ligands for catalytic studies. ROP catalysis is favored by reducing the bond strength of Zr–OR bond which is usually strong due to the high oxidation state of Zr(IV) ion. Multi-dentate ligands can donate electrons to metal through coordination, thereby weakening the metal–alkoxide bond.5 On the other hand, however, the pendant atoms on the hexadentate ligands is in direct competition with monomers which may result in a decreased polymerization rate. The difficulty is in identifying suitable pendant atoms that are capable of minimizing the competition with monomers by forming a labile coordination with Zr center. Herein, we report the syntheses of a series of hexadentate salan ligands, their associated kinetic studies, and their applications in ROP catalysis.
Results and discussion
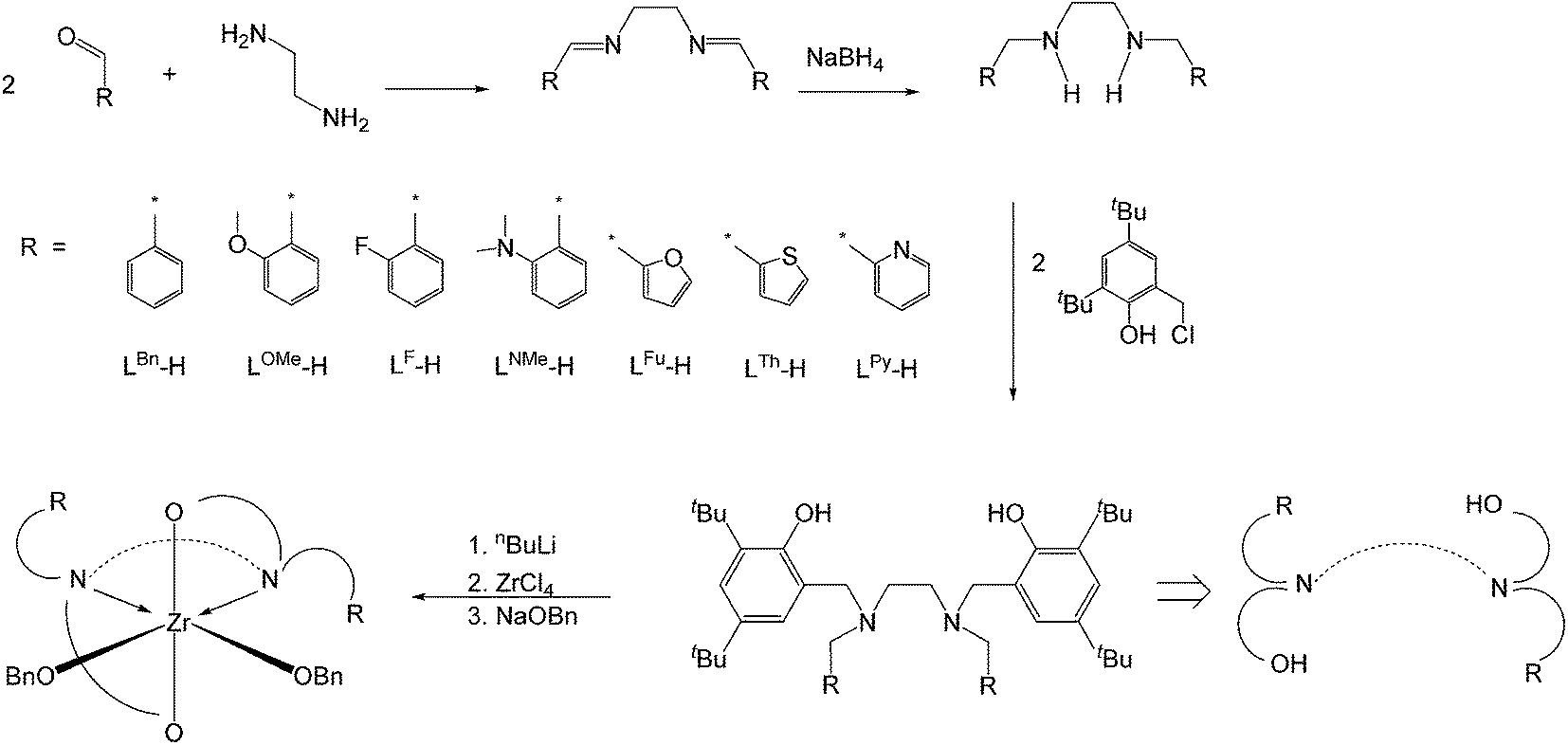
Synthesis and characterization of Zr complexes
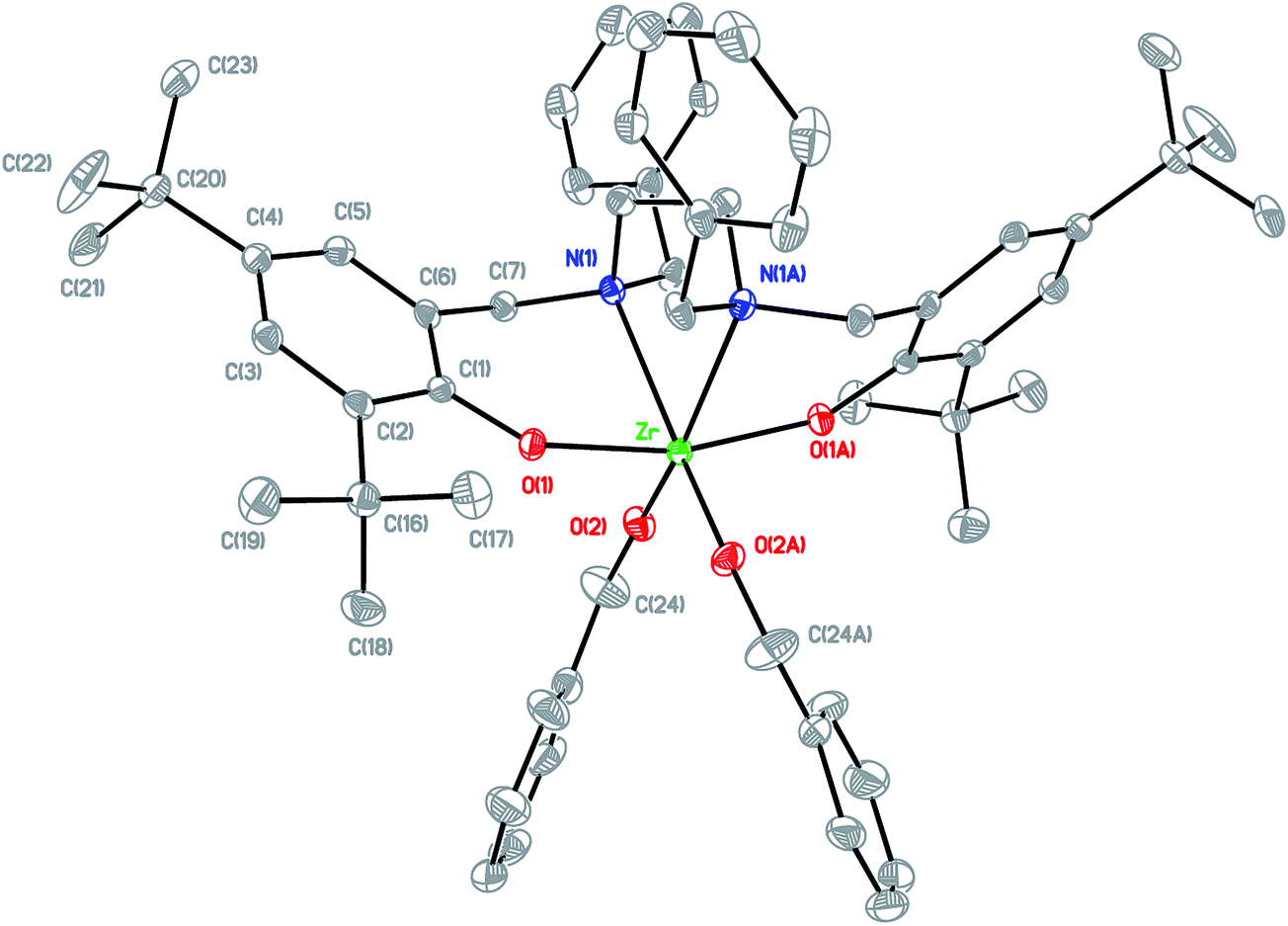
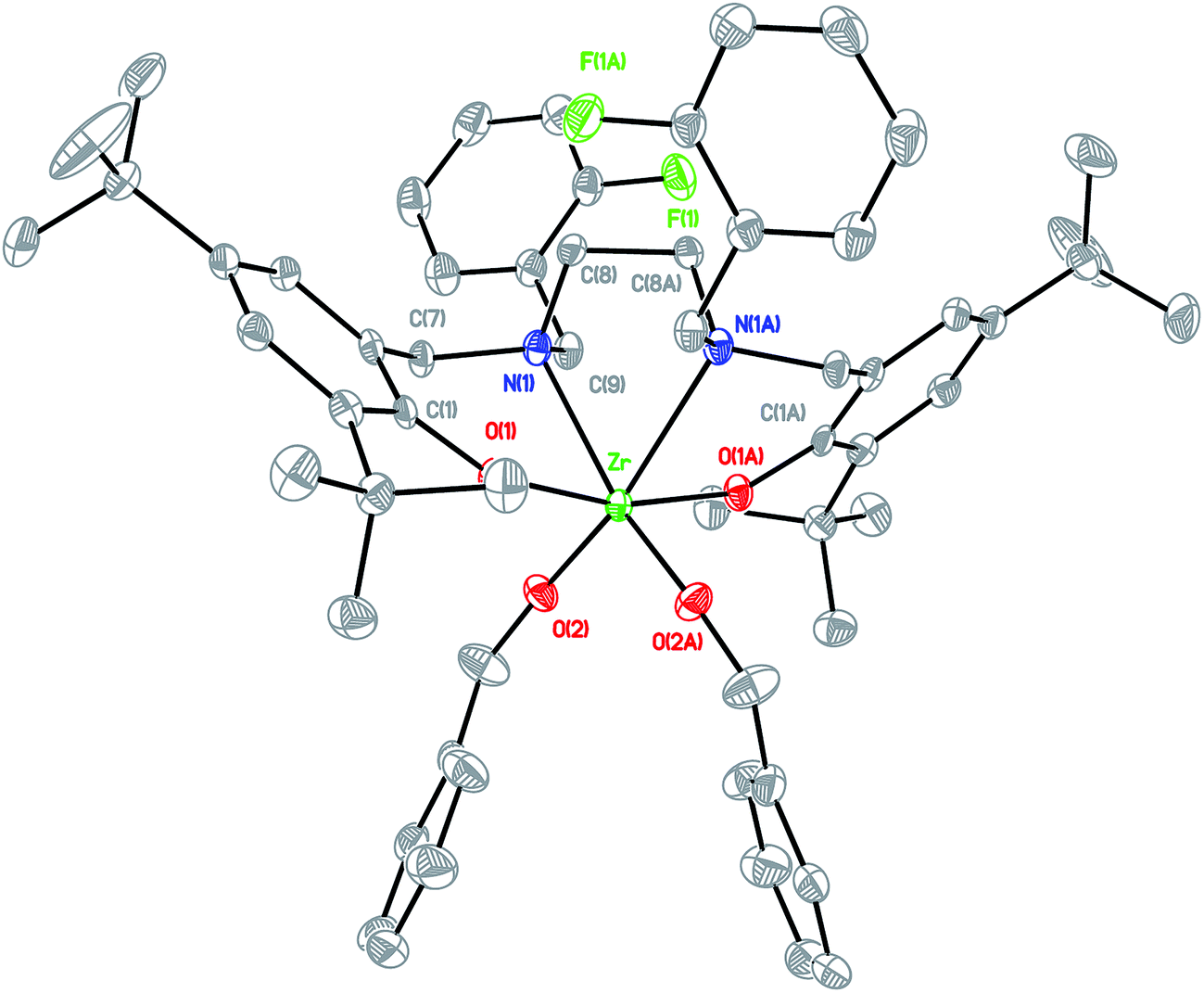
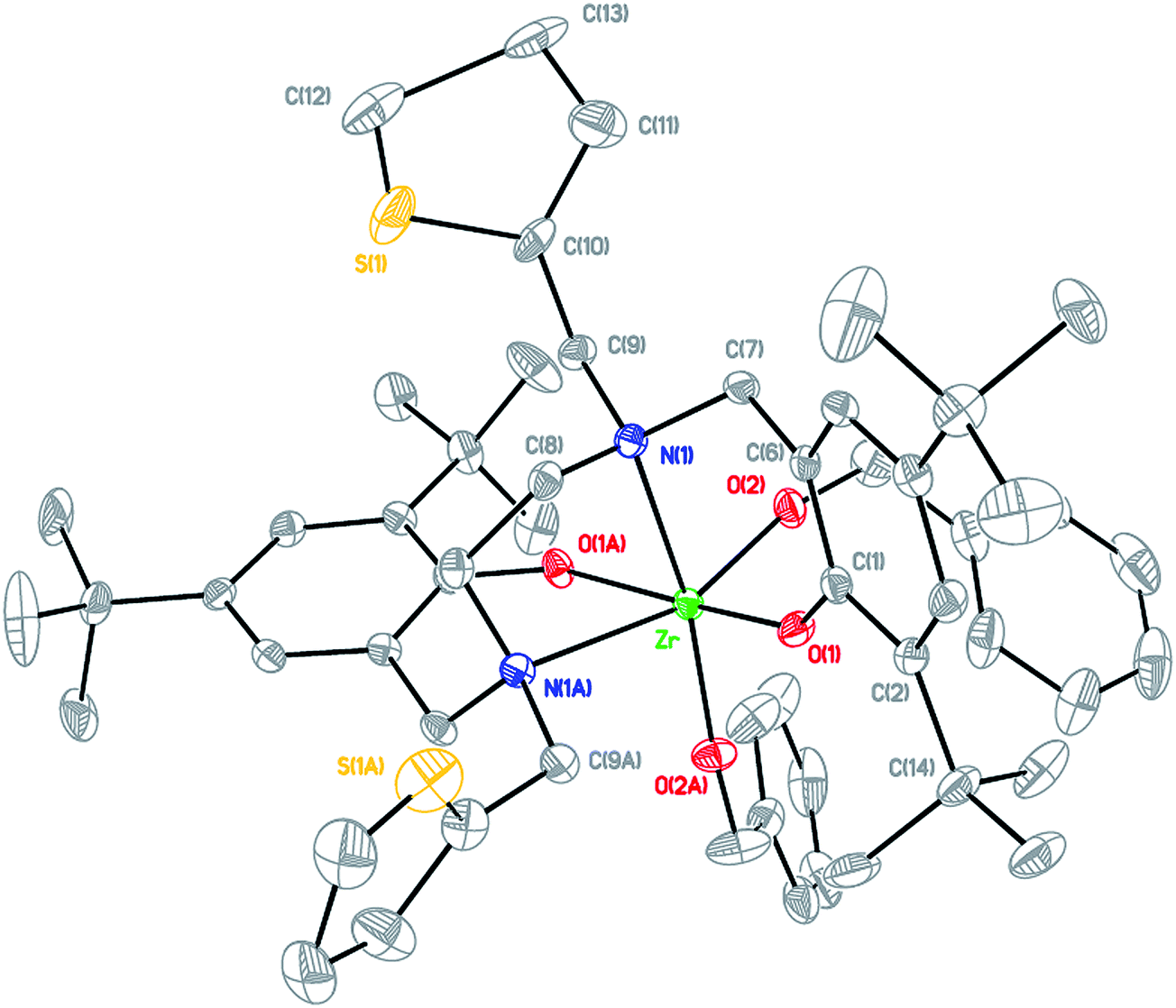
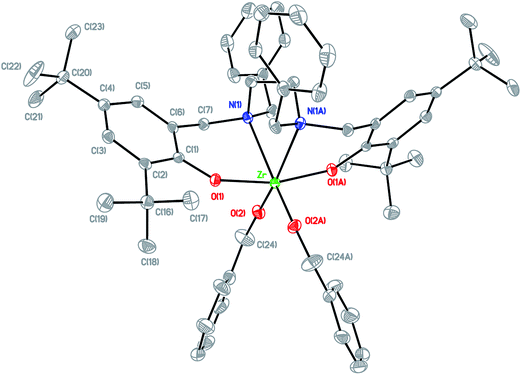
Arylaldehydes and ethyldiamine were condensed to produce diimines. Further reduction using NaBH4 followed by reaction with 2,4-di-tert-butyl-6-(chloromethyl)phenol offered a series of hexadentate salan ligands. All ligands were reacted with two equivalents of n-butyllithium in THF to produce a moderate yield of lithium compounds. These lithium complexes were then reacted with ZrCl4 to form zirconium dichloride complexes. Subsequent reaction with sodium benzyl alkoxide gave zirconium dibenzyl alkoxide complexes (Fig. 2). The structures of the final complexes were confirmed according to their 1H and 13C NMR spectra, elemental analyses, and X-ray crystallographic analyses. The X-ray structure of LBnZr(OBn)2 (Fig. 3) reveals that the zirconium complex is neutral, displaying two cis benzyl alkoxide and two trans phenolate groups (α-cis form). The axial angle of O(1)–Zr–O(1A) is 165.50(8)° and the equatorial angles between N(1)–Zr–N(1A), O(2)–Zr–N(1), and O(2)–Zr–O(2A) are 71.85(8), 91.87(6), and 105.88(9)°, respectively. The distances between the Zr atom and O(1), O(2), and N(1) are 2.0401(13), 1.9379(15), and 2.4684(17) Å, respectively, confirming that the structure is distorted from an ideal octahedral geometry. Moreover, the angles of C(24)–O(2)–Zr was 168.36(15)° with a strong π characteristic between zirconium and oxygen of benzyl alkoxide, which can be attributed to reduced bonding distance between the zirconium and oxygen of benzyl alkoxide. The X-ray structure of LFZr(OBn)2 (Fig. 4) and LThZr(OBn)2 (Fig. 5) present a geometry similar to that of LBnZr(OBn)2. In LFZr(OBn)2, the axial angles of O(1)–Zr–O(1A) is 165.64(15)o and the equatorial angles for N(1)–Zr–N(1A), O(2)–Zr–N(1), and O(2)–Zr–O(2A) are 72.28(14), 95.88(10), and 106.61(16)°, respectively. The distances between the Zr atom and O(1), O(2), and N(1) are 2.039(2), 1.942(2), and 2.472(3) Å, respectively, confirming that the structure was distorted from an ideal octahedral geometry. The angles of C(24)–O(2)–Zr is 168.8(3)°. In LThZr(OBn)2, the axial angles of O(1)–Zr–O(1A) is 164.69(8)° and the equatorial angles for N(1)–Zr–N(1A), O(2)–Zr–N(1), and O(2)–Zr–O(2A) are 72.41(8), 91.56(6), and 105.84(9)°, respectively. The distances between the Zr atom and O(1), O(2), and N(1) are 2.0408(14), 1.9391(14), and 2.4644(17) Å, respectively. Finally, the angles for C(22)–O(2)–Zr is 167.71(15)°.
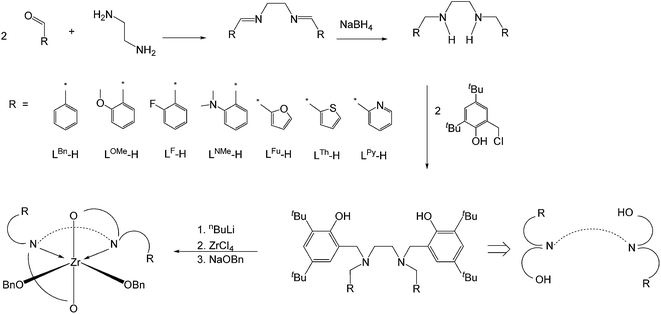 |
| | Fig. 2 Synthesis of bis(aminophenol) ligands and their Zr complexes. | |
 |
| | Fig. 3 Molecular structure of complex LBnZr(OBn)2 shown with 20% probability ellipsoids. CCDC deposition number: 1019663 (all of the hydrogen atoms were omitted for clarity). | |
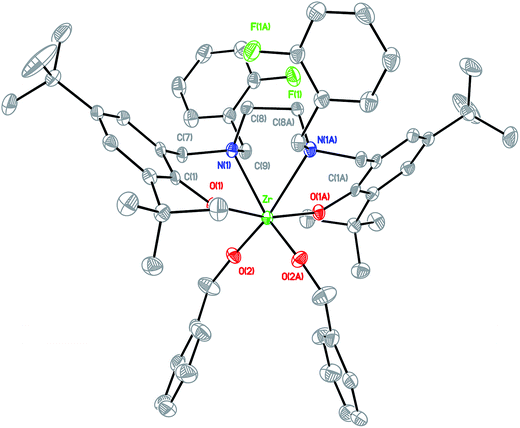 |
| | Fig. 4 Molecular structure of complex LFZr(OBn)2 shown with 20% probability ellipsoids. CCDC deposition number: 1019661 (all of the hydrogen atoms were omitted for clarity). | |
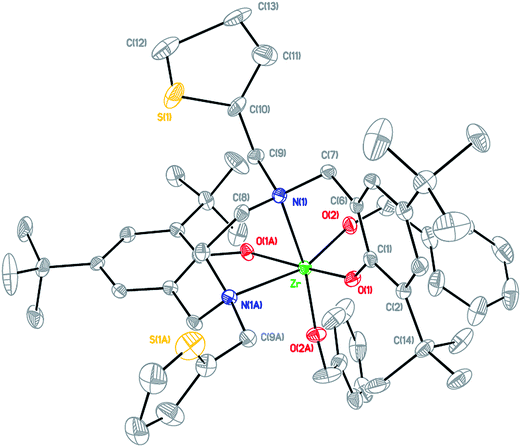 |
| | Fig. 5 Molecular structure of complex LThZr(OBn)2 shown with 20% probability ellipsoids. CCDC deposition number: 1019662 (all of the hydrogen atoms were omitted for clarity). | |
Polymerization of ε-caprolactone and L-lactide
We investigated the polymerizations of ε-caprolactone (CL) and L-lactide (LA) using zirconium complexes as initiators in toluene under nitrogen at 100 °C (Table 1). In Table 1, entries 1–7 for CL polymerization ([CL]/[Cat.] = 200), LFZr(OBn)2, LThZr(OBn)2, and LBnZr(OBn)2, (entries 1–3) showed greater activity than others and LPyZr(OBn)2 (entry 7) was least efficient. Their ability of polymer control was efficient with a limited polydispersity index (PDI) (PDI = 1.09–1.21) and anticipated molecular weight when two benzyl alkoxide were used as initiators. As shown in Table 1 (entries 8–14) for LA polymerization ([LA]/[Cat.] = 200), LFuZr(OBn)2, LOMeZr(OBn)2, and LPyZr(OBn)2 (entries 8–10) showed the greater activity and LThZr(OBn)2 (entry 14) was the least active catalyst. The Mn(NMR) of PLA catalyzed using LFuZr(OBn)2, LPyZr(OBn)2, and LBnZr(OBn)2 was inconsistent with Mn(GPC), perhaps due to the fact that transesterification was initiated by the catalysts for LA polymerization. Moreover, these Zr complexes appeared more active in the polymerization of CL than in the polymerization of LA. This trend is opposite to that of our previous findings related to Ti complexes.4
Table 1 Polymerization of CL and LA using each of the Zr complexes as an initiator at 100 °C
| Entry |
Catalyst LZr(OBn)2 |
Time (h) |
Conva |
Mn(Cal)b |
Mn(NMR)a |
Mn(GPC)c |
PDIc |
Obtained from 1H NMR analysis. Calculated from the molecular weight of monomer × [monomer]0/2[Cat]0 × conversion yield + Mw(OBn). Obtained from GPC analysis and calibration based on the polystyrene standard. Values in parentheses are the values obtained from GPC times 0.58 for PLA and 0.56 for PCL. Reaction condition: toluene (2 mL), [CL] = 5.0 M, [CL]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[Cat] = 200:1. Reaction condition: toluene (2 mL), [LA] = 5.0 M, [LA]:[Cat] = 200:1. :[Cat] = 200:1. Reaction condition: toluene (2 mL), [LA] = 5.0 M, [LA]:[Cat] = 200:1. |
| 1d |
LF |
4 |
99% |
11400 |
11000 |
11300 |
1.17 |
| 2d |
LTh |
4 |
99% |
10600 |
11800 |
11200 |
1.12 |
| 3d |
LFu |
4 |
99% |
11400 |
10900 |
15600 |
1.18 |
| 4d |
LBn |
4 |
82% |
9500 |
11200 |
9300 |
1.09 |
| 5d |
LNMe2 |
4 |
74% |
8500 |
11200 |
9100 |
1.16 |
| 6d |
LOMe |
4 |
54% |
6300 |
10400 |
9000 |
1.21 |
| 7d |
LPy |
4 |
42% |
4900 |
6700 |
4900 |
1.12 |
| 8e |
LFu |
48 |
89% |
12900 |
12500 |
5200 |
1.42 |
| 9e |
LOMe |
48 |
84% |
9700 |
9600 |
8000 |
1.02 |
| 10e |
LPy |
48 |
85% |
9800 |
15400 |
7300 |
1.12 |
| 11e |
LNMe2 |
48 |
75% |
9600 |
9800 |
7500 |
1.07 |
| 12e |
LF |
48 |
74% |
8500 |
11000 |
12700 |
1.03 |
| 13e |
LBn |
48 |
69% |
8000 |
8300 |
13000 |
1.06 |
| 14e |
LTh |
48 |
53% |
7700 |
8400 |
10400 |
1.03 |
To elucidate the catalytic behavior of these zirconium complexes involved in the polymerization of CL and LA, we conducted kinetic studies to determine the kobs (Table 2, Fig. S1 and S2, and Tables S1 andS2†). In Table 2, the trend of the activity of zirconium complexes with regard to polymerization in CDCl3 are similar to the trends observed in the polymerization activity in Table 1. However, the zirconium complexes used for polymerization in CL and LA presented precisely the opposite results. For example, the order of CL polymerization is LThZr(OBn)2 > LFZr(OBn)2 ≧ LFuZr(OBn)2 > LBnZr(OBn)2 > LNMe2Zr(OBn)2 > LOMeZr(OBn)2 ≧ LPyZr(OBn)2, whereas the order of LA polymerization is LPyZr(OBn)2 > LOMeZr(OBn)2 > LFuZr(OBn)2 > LNMe2Zr(OBn)2 > LBnZr(OBn)2 > LFZr(OBn)2 > LThZr(OBn)2. The differences between CL and LA are that the size of LA exceeds that of CL and the dipole moment of CL exceeds that of LA. Nevertheless it is difficult to explain the results according to these properties of CL and LA.
Table 2 Kinetic study of polymerization of ε-caprolactone and L-lactide using each of the Zr complexes as an initiator in a sealed NMR tube in CDCl3
| Catalyst LZr(OBn)2 |
CDCl3 |
| CL |
LA |
| Entry |
kobs |
Ranking |
kobs |
Ranking |
| LTh |
0.1162 (35) |
1 |
0.0265 (12) |
7 |
| LF |
0.0989 (67) |
2 |
0.0281 (17) |
6 |
| LFu |
0.0916 (73) |
3 |
0.3238 (153) |
3 |
| LBn |
0.0662 (18) |
4 |
0.0297 (15) |
5 |
| LNMe2 |
0.0424 (16) |
5 |
0.0337 (15) |
4 |
| LOMe |
0.0282 (10) |
6 |
0.3264 (85) |
2 |
| LPy |
0.0202 (3) |
7 |
1.1741 (363) |
1 |
Kinetic study of the polymerization of CL and LA catalyzed using LOMeZr(OBn)2
To rationalize our results related to the catalytic activity of these zirconium complexes in the polymerization of CL and LA, we conducted kinetic studies to establish the reaction order for monomer and catalysts. The experiments were performed using a ratio of [M]0/[LOMeZr(OBn)2] ([CL] = 0.2 M in 5 mL CH2Cl2 at room temperature and [LA] = 1.25 M in 1 mL CDCl3 at 100 °C) as shown in Tables S5, S6 and Fig. S5–S8.† Preliminary results indicate a first-order dependency on monomer ([CL] or [LA]) (Fig. S5 and S7†). By plotting ln kobs vs. ln [LOMeZr(OBn)2], we obtained an order of 2.17 for [LOMeZr(OBn)2], kp (propagation) values of 0.0015 for CL polymerization (Fig. S6†). By plotting kobs vs. [LOMeZr(OBn)2] under the assumption that the order of [LOMeZr(OBn)2] was 1, we obtained kp values of 1.6632 for LA polymerization (Fig. S6†). The polymerization of CL and LA using LOMeZr(OBn)2 demonstrated the following rate law:
| d[CL]/dt = 0.0015 × [LOMeZr(OiPr)2]2.17[CL]1 |
| d[LA]/dt = 1.6632 × [LOMeZr(OiPr)2]1[LA]1 |
Formulating an appropriate mechanism to explain the polymerization of CL and LA in accordance with the above kinetic data was challenging. It was necessary to rationalize the mechanism on the basis of the results observed during the catalytic activity of zirconium complexes in the polymerization of CL and LA. The order of [LOMeZr(OBn)2] is 2.17 for CL and 1 for LA. Therefore, one possible mechanism underlying CL polymerization would entail dinucleon8 from LOMeZr(OBn)2 aggregation acting as the real active species. In LA polymerization, this would imply the mononuclear form of LOMeZr(OBn)2. In addition, CL and LA polymerizations using zirconium complexes as catalysts requires no induction period, unlike the previously reported case with titanium complexes.4 One reason for this may be that the zirconium complexes with a six-coordinate metal center do not have to transform into other species in order to be coordinated with CL or LA since the maximum coordination number of Zr ion is eight.
Conclusions
This study synthesized a series of zirconium complexes bearing salan ligands to catalyze the polymerization of CL and LA. The polymerization rate of CL and LA showed opposing trends, according to the pendent group. Among the zirconium complexes, the thiophen-2-yl methyl group was most effective in enhancing the polymerization rate of CL, whereas the pyridine-2-ylmethyl group was most effective in polymerization of LA. Kinetic studies indicated a first-order dependency on [CL] and [LA] respectively. Polymerization proceeded with second-order dependence on [LOMeZr(OBn)2] for CL but with first-order dependence on [LOMeZr(OBn)2] for LA. These results revealed that the chelating groups influenced the polymerization activity of zirconium complexes. However, the effect of chelation differ between CL and LA polymerization.
Experimental section
Standard Schlenk techniques and a N2-filled glovebox were used throughout the isolation and treatment of all compounds. Solvents, ε-caprolactone, L-lactide, and deuterated solvents were purified prior to use. 2,4-Di-tert-butylphenol, sodium borohydride, formaldehyde (37 wt% sol. in water), triethylamine, thionyl chloride, ethylenediamine anhydrous, benzaldehyde, picolinaldehyde, 2-methoxybenzaldehyde, 2-fluorobenzaldehyde, thiophene-2-carbaldehyde, furan-2-carbaldehyde, titanium(IV) isopropoxide, sodium hydride, deuterated chloroform, L-lactide, and ε-caprolactone were purchased from Acros. Benzyl alcohol was purchased from Alfa. 1H and 13C NMR spectra were recorded on a Varian Gemini2000-200 (200 MHz for 1H and 50 MHz for 13C) spectrometer with chemical shifts given in ppm from the internal TMS or center line of CDCl3. Microanalyses were performed using a Heraeus CHN-O-RAPID instrument. GPC measurements were performed on a Jasco PU-2080 PLUS HPLC pump system equipped with a differential Jasco RI-2031 PLUS refractive index detector using THF (HPLC grade) as an eluent (flow rate 1.0 mL min−1, at 40 °C). The chromatographic column was JORDI Gel DVB 103 Å, and the calibration curve was made by primary polystyrene standards to calculate Mn(GPC). 2,4-Di-tert-butyl-6-(chloromethyl)phenol,6 LBn-H2,4 LOMe-H2, LF-H2, LFu-H2, LTh-H2, and 2-(dimethylamino)benzaldehyde7 were prepared following literature procedures.
Synthesis of N,N′-bis(2-dimethylaminobenzyl)-N,N′-bis[(3,5-di-tert-butyl-2-hydroxyphenyl)-methylene]-1,2-diaminoethane (LNMe2-H2)
A mixture of ethylenediamine (6.01 g, 100 mmol) and 2-(dimethylamino)benzaldehyde (24.80 g, 200 mmol) was refluxed for one day in ethanol (150 mL). The reaction solution was cooled down in ice bath and sodium borohydride (7.57 g, 200 mmol) was transferred to the solution slowly. After 1 h, the solution was refluxed again for a day. Volatile materials were removed under vacuum to yield yellow oil. The oil was dissolved in CH2Cl2 (200 mL) and the solution was washed with water (2/200 mL). After solvent removal under reduced pressure, white powder was obtained. The white powder was set with 2,4-di-tert-butyl-6-(chloromethyl)phenol (53.55 g, 210 mmol) and NEt3 (28 mL, 200 mmol) in 400 mL ethanol and refluxed for one month. Volatile materials were removed under vacuum to yield yellow oil. The oil was dissolved in CH2Cl2 (200 mL) and the solution was washed with water (2/200 mL) and several drops of HCl (37%). The yellow oil was obtained when CH2Cl2 was removed and ethanol (250 mL) was added to dissolve the oil. The white powder was obtained and filtered after 10 day at −20 °C. Yield: 48.84 g (64%). 1H NMR (CDCl3, 200 MHz): δ 10.84 (2H, s, ArOH), 7.23–6.78 (12H, m, ArH), 3.60 (4H, s, NCH2PhN(CH3)2), 3.59 (4H, s, NCH2Ar), 2.63 (4H, s, NCH2CH2N), 2.50 (12H, PhN(CH3)2), 1.39 (18H, s, ArC(CH3)3), 1.24 (18H, s, ArC(CH3)3). 13C NMR (CDCl3, 50 MHz): δ 154.08, 153.58, 140.18, 135.36, 131.77, 130.91, 128.17, 123.50, 123.48, 122.68, 121.41, 119.57 (Ar), 58.98 (NCH2PhN(CH3)2), 53.74 (NCH2Ar), 50.32 (NCH2CH2N), 45.16 (PhN(CH3)2), 34.81 (ArC(CH3)3), 34.06 (ArC(CH3)3), 31.70 (ArC(CH3)3), 29.55 (ArC(CH3)3). Elemental analysis (C50H74N4O2) found: N, 7.55%; C, 78.39%; H, 9.44%. Anal. calcd: N, 7.34%; C, 78.69%; H, 9.77%. ESI-MS(+) m/z calcd = 763.15. Found: 763.49.
Synthesis of N,N′-bis(pyridin-2-methylbenzyl)-N,N′-bis[(3,5-di-tert-butyl-2-hydroxyphenyl)-methylene]-1,2-diaminoethane (LPy-H2)
Synthetic procedures were similar to that of LNMe2-H2 except pyridine-2-methylbenzaldehyde was used in place of 2-(dimethylamino)benzaldehyde. 1H NMR (CDCl3, 200 MHz): δ 10.46 (2H, s, ArOH), 8.50 (2H, d, J = 4.8 Hz, PyrH), 7.59–6.79 (10H, m, ArH, PyrH), 3.70 (8H, s, NCH2Pyr), 3.59 (4H, s, NCH2Ar), 2.81 (4H, s, NCH2CH2N), 1.39 (18H, s, ArC(CH3)3), 1.25 (18H, s, ArC(CH3)3). 13C NMR (CDCl3, 50 MHz): δ 157.47, 153.82, 149.01, 140.53, 136.52, 135.60, 123.92, 123.62, 122.98, 122.26, 121.17 (Ar), 59.43 (NCH2Pyr), 59.01 (NCH2Ar), 50.47 (NCH2CH2N), 34.85 (ArC(CH3)3), 34.09 (ArC(CH3)3), 31.67 (ArC(CH3)3), 29.56 (ArC(CH3)3). Elemental analysis (C44H62N4O2) found: N, 3.21%; C, 77.59%; H, 9.29%. Anal. calcd: N, 8.25%; C, 77.83%; H, 9.20%. ESI-MS(+) m/z calcd = 678.49. Found: 679.31.
Synthesis of LBnZr(OBn)2
A mixture of LBn-H2 (6.77 g, 10 mmol) and n-butyllithium (8.00 mL, 2.5 M) in THF (40 mL) was stirred for 3 h. Volatile materials were removed under vacuum and then zirconium(IV) chloride (2.29 g 10 mmol) dissolved in THF (40 mL) was added. After stirring for one day, the mixture was reacted with sodium benzyl alkoxide that was synthesized from sodium hydride (0.48 g, 20 mmol) and benzyl alcohol (2.16 g, 20 mmol) for another day. Volatile materials were removed under vacuum again and toluene (20 mL) was added to form a suspension. The sodium and lithium salts were removed by filtration and yellow powder was obtained under vacuum. It was washed with hexane (30 mL) to afford final product as light yellow powder. Yield: 6.17 g (63%). 1H NMR (CDCl3, 200 MHz): δ 7.44–6.01 (24H, m, ArH), 5.29 (4H, s, OCH2Ph), 4.22 (2H, dd, J = 14.2 Hz, NCH2Ph), 4.06 (2H, dd, J = 13.4 Hz, NCH2Ar), 4.22 (2H, dd, J = 14.2 Hz, NCH2Ph), 3.91 (2H, dd, J = 14.2 Hz, NCH2Ph), 3.32 (2H, dd, J = 13.4 Hz, NCH2Ar), 2.70 (2H, dd, J = 9.8 Hz, NCH2CH2N), 2.38 (2H, dd, J = 9.8 Hz, NCH2CH2N), 1.50 (18H, s, ArC(CH3)3), 1.26 (18H, s, ArC(CH3)3). 13C NMR (CDCl3, 50 MHz): δ 158.04, 143.80, 138.89, 136.59, 132.35, 131.40, 128.17, 127.97, 126.44, 126.34, 126.22, 124.71, 124.05, 123.42 (Ar), 72.25 (OCH2Ph), 59.13 (NCH2Ar), 58.33 (NCH2Ph), 45.93 (NCH2CH2N), 35.19 (ArC(CH3)3), 34.13 (ArC(CH3)3), 31.79 (ArC(CH3)3), 30.13 (ArC(CH3)3). Elemental analysis (C61H79N2O4Zr) found: N, 2.87%; C, 73.58%; H, 8.05%. Anal. calcd: N, 2.86%; C, 73.50%; H, 7.81%. Mp: 224 °C.
Synthesis of LOMeZr(OBn)2
Synthetic procedures were similar to that for LBnZr(OBn)2 except LOMe-H2 was used in place of LBn-H2. Yield: 8.84 g (85%). 1H NMR (CDCl3, 200 MHz): δ 7.47–6.62 (22H, m, ArH), 5.34 (4H, s, OCH2Ph), 4.45 (2H, dd, J = 13.8 Hz, NCH2PhOMe), 4.27 (2H, dd, J = 13.0 Hz, NCH2Ar), 3.97 (2H, dd, J = 13.8 Hz, NCH2PhOMe), 3.39 (2H, dd, J = 13.0 Hz, NCH2Ar), 3.22 (6H, s, NCH2PhOCH3), 3.15 (2H, dd, J = 9.2 Hz, NCH2CH2N), 1.96 (2H, dd, J = 9.2 Hz, NCH2CH2N), 1.48 (18H, s, ArC(CH3)3), 1.22 (18H, s, ArC(CH3)3). 13C NMR (CDCl3, 50 MHz): δ 189.86, 159.36, 157.96, 144.21, 138.34, 136.61, 134.85, 129.72, 127.86, 126.19, 125.98, 124.65, 123.95, 123.65, 120.77, 120.36, 120.25, 111.46 (Ar), 71.96 (OCH2Ph), 59.35 (NCH2PhOMe), 55.32 (OCH3), 51.43 (NCH2PhOMe), 45.22 (NCH2CH2N), 35.13 (ArC(CH3)3), 34.08 (ArC(CH3)3), 31.84 (ArC(CH3)3), 30.10 (ArC(CH3)3). Elemental analysis (C63H83N2O6Zr) found: N, 2.95%; C, 72.03%; H, 7.80%. Anal. calcd: N, 2.69%; C, 71.57%; H, 7.75%. Mp: 207 °C.
Synthesis of LFZr(OBn)2
Synthetic procedures were similar to that for LBnZr(OBn)2 except LF-H2 was used in place of LBn-H2. Yield: 7.01 g (69%). 1H NMR (CDCl3, 200 MHz): δ 7.43–6.60 (22H, m, ArH), 5.31 (4H, s, OCH2Ph), 4.31 (2H, dd, J = 10.0 Hz, OCH2PhF), 4.21 (2H, dd, J = 13.0 Hz, NCH2Ar), 4.15 (2H, dd, J = 10.0 Hz, NCH2PhF), 3.22 (2H, dd, J = 13.0 Hz, NCH2Ar), 2.96 (2H, dd, J = 10.6 Hz, NCH2CH2N), 2.24 (2H, dd, J = 10.6 Hz, NCH2CH2N), 1.50 (18H, s, ArC(CH3)3), 1.24 (18H, s, ArC(CH3)3). 13C NMR (CDCl3, 50 MHz): δ 157.68, 143.79, 138.90, 136.90, 136.45, 134.84, 130.56, 130.39, 127.92, 126.34, 126.16, 124.78, 123.91, 123.15, 118.95, 118.60, 116.90, 115.63 (Ar), 72.21 (OCH2Ph), 59.27 (NCH2PhF), 51.20 (NCH2PhF), 45.73 (NCH2CH2N), 35.11 (ArC(CH3)3), 34.08 (ArC(CH3)3), 31.73 (ArC(CH3)3), 30.09 (ArC(CH3)3). Elemental analysis (C61H77N2F2O4Zr) found: N, 2.60%; C, 71.13%; H, 7.35%. Anal. calcd: N, 2.76%; C, 70.90%; H, 7.34%. Mp: 218 °C.
Synthesis of LNMe2Zr(OBn)2
Synthetic procedures were similar to that for LBnZr(OBn)2 except LNMe2-H2 was used in place of LBn-H2. Yield: 8.74 g (82%). 1H NMR (CDCl3, 400 MHz): δ 7.48–6.58 (22H, m, ArH), 5.36 (4H, s, OCH2Ph), 4.52 (2H, dd, J = 14.0 Hz, NCH2PhNMe2), 4.22 (2H, dd, J = 12.8 Hz, NCH2Ar), 3.97 (2H, dd, J = 14.0 Hz, NCH2PhNMe2), 3.39 (2H, dd, J = 12.8 Hz, NCH2Ar), 3.15 (2H, dd, J = 9.2 Hz, NCH2CH2N), 1.98 (12H, s, NCH2PhN(CH3)2), 1.96 (2H, dd, J = 9.2 Hz, NCH2CH2N), 1.48 (18H, s, ArC(CH3)3), 1.22 (18H, s, ArC(CH3)3). 13C NMR (CDCl3, 50 MHz): δ 157.94, 155.74, 144.22, 138.53, 136.40, 135.29, 129.40, 127.87, 127.20, 126.21, 126.07, 126.03, 124.94, 124.24, 123.65, 123.59, 120.93 (Ar, Ph), 71.99 (OCH2Ph), 58.95 (NCH2 BnN(CH3)2), 52.25 (NCH2 Ar), 44.78 (NCH2CH2N), 44.46 (BnN(CH3)2),35.16 (ArC(CH3)3), 34.07 (ArC(CH3)3), 31.82 (ArC(CH3)3), 30.08 (ArC(CH3)3). Elemental analysis (C65H89N4O4Zr) found: N, 3.57%; C, 72.27%; H, 7.84%. Anal. calcd: N, 5.25%; C, 72.07%; H, 8.13%. Mp: 178 °C.
Synthesis of LPyZr(OBn)2
Synthetic procedures were similar to that for LBnZr(OBn)2 except LPy-H2 was used in place of LBn-H2. Yield: 7.27 g (74%). 1H NMR (CDCl3, 400 MHz): δ 9.03 (PyrH), 7.60–6.60 (20H, m, ArH), 5.46 (4H, d, J = 4 Hz, OCH2Ph), 4.61 (2H, dd, J = 14.8 Hz, NCH2Py), 4.38 (2H, dd, J = 13.2 Hz, NCH2Ar), 3.57 (2H, dd, J = 14.8 Hz, NCH2Py), 3.43 (2H, dd, J = 13.2 Hz, NCH2Ar), 3.11 (2H, dd, J = 13.2 Hz, NCH2CH2N), 2.40 (2H, dd, J = 13.2 Hz, NCH2CH2N), 1.29 (18H, s, ArC(CH3)3), 1.24 (18H, s, ArC(CH3)3). 13C NMR (CDCl3, 100 MHz): δ 156.57, 150.54, 145.58, 138.48, 137.10, 136.80, 127.63, 127.60, 126.34, 126.23, 126.13, 125.51, 125.45, 124.74, 123.83, 123.25, 122.43 (Ar, Pyr), 71.18 (OCH2Ph), 59.85 (NCH2Pyr), 50.29 (NCH2Ar), 48.24 (NCH2CH2N), 34.80 (ArC(CH3)3), 34.02 (ArC(CH3)3), 30.52 (ArC(CH3)3), 29.81 (ArC(CH3)3). Elemental analysis (C59H77N4O4Zr) found: 5.60%; C, 69.60%; H, 8.03%. Anal. calcd: N, 5.70%; C, 70.91%; H, 7.59%. Mp: 168 °C.
Synthesis of LThZr(OBn)2
Synthetic procedures were similar to that for LBnZr(OBn)2 except LTh-H2 was used in place of LBn-H2. Yield: 6.84 g (69%). 1H NMR (CDCl3, 200 MHz): δ 7.36–7.15 (14H, m, ArH), 6.89–6.58 (6H, m, ThioH), 5.21 (4H, s, OCH2Ph), 4.46 (2H, dd, J = 15.8 Hz, NCH2Th), 4.22 (2H, dd, J = 15.8 Hz, NCH2Th), 4.03 (2H, dd, J = 13.0 Hz, NCH2Ar), 3.77 (2H, dd, J = 14.8 Hz, NCH2CH2N), 3.55 (2H, dd, J = 13.2 Hz, NCH2Ar), 2.71 (2H, dd, J = 13.2 Hz, NCH2CH2N), 1.50 (18H, s, ArC(CH3)3), 1.28 (18H, s, ArC(CH3)3). 13C NMR (CDCl3, 50 MHz): δ 158.15, 143.51, 139.06, 136.58, 133.09, 130.88, 127.98, 126.96, 126.89, 126.43, 126.29, 124.88, 124.16, 123.18 (Ar), 72.28 (OCH2Ph), 58.68 (NCH2Th), 52.39 (NCH2Ar), 47.08 (NCH2CH2N), 35.17 (ArC(CH3)3), 34.16 (ArC(CH3)3), 31.81 (ArC(CH3)3), 30.11 (ArC(CH3)3). Elemental analysis (C57H75N2O4S2Zr) found: 2.74%; C, 67.73%; H, 6.95%. Anal. calcd: N, 2.82%; C, 67.77%; H, 7.31%. Mp: 174 °C.
Synthesis of LFuZr(OBn)2
Synthetic procedures were similar to that for LBnZr(OBn)2 except except LFu-H2 was used in place of LBn-H2. Yield: 3.45 g (36%). 1H NMR (CDCl3, 200 MHz): δ 7.37–7.14 (14H, m, ArH), 6.70, 6.32, 6.03 (6H, m, FuH), 5.20 (4H, s, OCH2Ph), 4.26 (2H, dd, J = 15.8 Hz, NCH2Fu), 4.06 (2H, dd, J = 13.2 Hz, NCH2Ar), 4.00 (2H, dd, J = 15.8 Hz, NCH2Fu), 3.45 (2H, dd, J = 13.2 Hz, NCH2Ar), 2.84 (2H, dd, J = 10.2 Hz, NCH2CH2N), 2.45 (2H, dd, J = 10.2 Hz, NCH2CH2N), 1.50 (18H, s, ArC(CH3)3), 1.29 (18H, s, ArC(CH3)3). 13C NMR (CDCl3, 50 MHz): δ 157.85, 148.18, 143.60, 143.29, 138.90, 136.52, 127.92, 126.30, 126.17, 124.88, 123.94, 123.15, 112.71, 110.32 (Ar, Ph, Furan), 72.13 (OCH2Ph), 59.37 (NCH2Furan), 50.24 (NCH2 Ar), 46.81 (NCH2CH2N), 35.14 (ArC(CH3)3), 34.13 (ArC(CH3)3), 31.79 (ArC(CH3)3), 30.06 (ArC(CH3)3). Elemental analysis (C57H75N2O6Zr) found: 2.76%; C, 69.91%; H, 7.74%. Anal. calcd: N, 2.92%; C, 70.03%; H, 7.56%. Mp: 122 °C.
General procedures for the polymerization of CL
A typical polymerization procedure was exemplified by the synthesis of entry 6 (Table 1) using complex LOMeZr(OBn)2 as a catalyst. The polymerization conversion was analyzed by 1H NMR spectroscopic studies. Toluene (2.0 mL) was added to a mixture of complex LOMeZr(OBn)2 (0.05 mmol) and ε-caprolactone (1.14 g, 10 mmol) at 100 °C. After the solution was stirred for 4 h, the reaction was quenched by adding a drop of ethanol. Then the polymer was precipitated as white solid by pouring into n-hexane (30.0 mL). The white solid was redissolved in CH2Cl2 (5.0 mL) and then n-hexane (70.0 mL) added to give white crystalline solid. Yield: 0.62 g (54%).
Acknowledgements
This study is supported by Kaohsiung Medical University “Aim for the top 500 universities grant” under Grant no. KMU-DT103007, NSYSU-KMU JOINT RESEARCH PROJECT (NSYSU KMU 103-I004), and the Ministry of Science and Technology (Grant NSC 101-2113-M-037 -009). We thank Center for Research Resources and Development at Kaohsiung Medical University for the instrumentation and equipment support.
References
-
(a) C.-K. Huang, C.-L. Lo, H.-H. Chen and G.-H. Hsiue, Adv. Funct. Mater., 2007, 17, 2291–2297 CrossRef CAS;
(b) L. Ilario, I. Francolini, A. Martinelli and A. Piozzi, Macromol. Rapid Commun., 2007, 28, 1900–1904 CrossRef;
(c) S. Slomkowski, Macromol. Symp., 2007, 253, 47–58 CrossRef CAS;
(d) W. Y. Ip, S. Gogolewski and K. Tsui, Eur. Cells Mater., 2003, 5, 7 Search PubMed;
(e) R. L. Simpson, F. E. Wiria, A. A. Amis, C. K. Chua, K. F. Leong, U. N. Hansen, M. Chandrasekaran and M. W. Lee, J. Biomed. Mater. Res., Part B, 2007, 69, 17 Search PubMed;
(f) E. T. H. Vinka, K. R. Rábagob, D. A. Glassnerb and P. R. Gruberb, Polym. Degrad. Stab., 2003, 80, 403–419 CrossRef;
(g) D. Lickorisha, L. Guana and J. E. Daviesa, Biomaterials, 2007, 28, 1495–1502 CrossRef PubMed;
(h) R. E. Drumright, P. R. Gruber and D. E. Henton, Adv. Mater., 2000, 12, 1841–1846 CrossRef CAS;
(i) C.-S. Ha Jr and J. A. Gardella, Chem. Rev., 2005, 105, 4205–4232 CrossRef PubMed;
(j) D. Farrar, Bus. Brief.: Med. Device Manuf. Technol., 2005, 1–4 Search PubMed.
-
(a) C. K. Williams, L. E. Breyfogle, S. K. Choi, W. Nam, V. G. Young Jr, M. A. Hillmyer and W. B. Tolman, J. Am. Chem. Soc., 2003, 125, 11350–11359 CrossRef CAS PubMed;
(b) A. K. Sutar, T. Maharana, S. Dutta, C.-T. Chen and C.-C. Lin, Chem. Soc. Rev., 2010, 39, 1724–1746 RSC;
(c) M. H. Chisholm, N. W. Eilerts, J. C. Huffman, S. S. Iyer, M. Pacold and K. Phomphrai, J. Am. Chem. Soc., 2000, 12, 11845–11854 CrossRef;
(d) D. J. Darensbourg, W. Choi, O. Karroonnirun and N. Bhuvanesh, Macromolecules, 2008, 41, 3493–3502 CrossRef CAS;
(e) B. J. O'Keefe, L. E. Breyfogle, M. A. Hillmyer and W. B. Tolman, J. Am. Chem. Soc., 2002, 124, 4384–4393 CrossRef PubMed;
(f) A. P. Dove, V. C. Gibson, E. L. Marshall, H. S. Rzepa, A. J. P. White and D. J. Williams, J. Am. Chem. Soc., 2006, 128, 9834–9843 CrossRef CAS PubMed;
(g) K. Majerska and A. Duda, J. Am. Chem. Soc., 2004, 126, 1026–1027 CrossRef CAS PubMed.
-
(a) A. J. Chmura, M. G. Davidson, C. J. Frankis, M. D. Jones and M. D. Lunn, Chem. Commun., 2008, 44, 1293–1295 RSC;
(b) B. J. Jeffery, E. L. Whitelaw, D. Garcia-Vivo, J. A. Stewart, M. F. Mahon, M. G. Davidson and M. D. Jones, Chem. Commun., 2011, 47, 12328–12330 RSC;
(c) A. J. Chmura, M. G. Davidson, M. D. Jones, M. D. Lunn, M. F. Mahon, A. F. Johnson, P. Khunkamchoo, S. L. Roberts and S. S. F. Wong, Macromolecules, 2006, 39, 7250–7257 CrossRef CAS;
(d) S. Gendler, S. Segal, I. Goldberg, Z. Goldschmidt and M. Kol, Inorg. Chem., 2006, 45, 4783–4790 CrossRef CAS PubMed;
(e) M. Hu, M. Wang, H. Zhu, L. Zhang, H. Zhang and L. Sun, Dalton Trans., 2010, 39, 4440–4446 RSC;
(f) M. Hu, M. Wang, P. Zhang, K. Jin, Y. Chen and L. Sun, Polym. Bull., 2012, 68, 1789–1799 CrossRef CAS;
(g) T. K. Saha, V. Ramkumar and D. Chakraborty, Inorg. Chem., 2011, 50, 2720–2722 CrossRef CAS PubMed;
(h) E. L. Whitelaw, M. D. Jones and M. F. Mahon, Inorg. Chem., 2010, 49, 7176–7181 CrossRef CAS PubMed;
(i) E. L. Whitelaw, M. G. Davidson and M. D. Jones, Chem. Commun., 2011, 47, 10004–10006 RSC;
(j) A. Stopper, J. Okuda and M. Ko, Macromolecules, 2012, 45, 698–704 CrossRef CAS;
(k) A. Sauer, J.-C. Buffet, T. P. Spaniol, H. Nagae, K. Mashima and J. Okuda, Inorg. Chem., 2012, 51, 5764–5770 CrossRef CAS PubMed;
(l) J.-C. Buffet and J. Okuda, Chem. Commun., 2011, 47, 4796–4798 RSC;
(m) C. Romain, B. Heinrich, S. B. Laponnaz and S. Dagorne, Chem. Commun., 2012, 48, 2213–2215 RSC.
- H.-W. Ou, H.-Y. Chen, H.-C. Tseng, M.-W. Hsiao, Y.-L. Chang, N.-Y. Jheng, Y.-C. Lai, T.-Y. Shih, Y.-T. Lin and H.-Y. Chen, J. Mol. Catal. A: Chem., 2014, 394, 97–104 CrossRef CAS PubMed.
- H.-Y. Chen, M.-Y. Liu, A. K. Sutar and C.-C. Lin, Inorg. Chem., 2010, 49, 665–674 CrossRef CAS PubMed.
- M. Lanznaster, H. P. Hratchian, M. J. Heeg, L. M. Hryhorczuk, B. R. McGarvey, H. B. Schlegel and C. N. Verani, Inorg. Chem., 2006, 45, 955–957 CrossRef CAS PubMed.
- A. Gao, Y. Mu, J. Zhang and W. Yao, Eur. J. Inorg. Chem., 2009, 3613–3621 CrossRef CAS.
- T. R. Forder, M. F. Mahon, M. G. Davidson, T. Woodmanc and M. D. Jones, Dalton Trans., 2014, 43, 12095–12099 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Polymer characterization data and details of the kinetic study are available. CCDC 1019661, 1019662 and 1019663. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ra13236j |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.