
Aligning 3D nanofibrous networks from self-assembled phenylalanine nanofibers†
Abstract
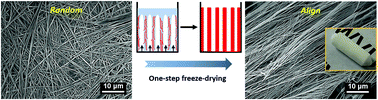
Self-assembled synthetic materials are typically disordered, and controlling the alignment of such materials at the nanometer scale may be important for a variety of biological applications. In this study, we have applied directional freeze-drying, for the first time, to develop well aligned three dimensional (3D) nanofibrous materials using amino acid like L-phenylalanine (Phe). 3D free-standing Phe nanofibrous monoliths have been successfully prepared using directional freeze-drying, and have presented a unique hierarchical structure with well-aligned nanofibers at the nanometer scale and an ordered compartmental architecture at the micrometer scale. We have found that the physical properties (e.g. nanofiber density and alignment) of the nanofibrous materials could be tuned by controlling the concentration and pH of the Phe solution and the freezing temperature. Moreover, the same strategy (i.e. directional freeze-drying) has been successfully applied to assemble peptide nanofibrous materials using a dipeptide (i.e. diphenylalanine), and to assemble Phe-based nanofibrous composites using polyethylenimine and poly(vinyl alcohol). The tunability of the nanofibrous structures together with the biocompatibility of Phe may make these 3D nanofibrous materials suitable for a variety of applications, including biosensor templates, tissue scaffolds, filtration membranes, and absorbents. The strategy reported here is likely applicable to create aligned nanofibrous structures using other amino acids, peptides, and polymers.
 Please wait while we load your content...
Please wait while we load your content...

