
Computational aspects of hydroformylation
Tamás Kégl*
Department of Inorganic Chemistry, University of Pécs and János Szentágothai Science Center, MTA-PTE Research Group for Selective Chemical Syntheses, H-7624 Pécs, Hungary. E-mail: tkegl@gamma.ttk.pte.hu
First published on 4th December 2014
Abstract
The influence of transition metal complexes as catalysts upon the activity and selectivity of hydroformylation reactions has been extensively investigated during the last decades. Nowadays computational chemistry is an indispensable tool for elucidation of reaction mechanisms and for understanding the various aspects which govern the outcome of catalytic reactions. This review attempts to survey the recent literature concerning computational studies on hydroformylation and theoretical coordination chemistry results related to hydroformylation.
1. Introduction
The hydroformylation or oxo reaction, discovered by Otto Roelen1 is the transition metal mediated addition of carbon monoxide and dihydrogen to the double bond of an alkene. It is one of the most versatile methods for the functionalization of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds and can be considered consequently as a very robust synthetic tool. The primary products of the exothermic reaction are aldehyde isomers. If the carbon atoms of the double bond are equivalent, only one aldehyde is formed, as depicted in Scheme 1(a). Substituted alkenes afford at least two aldehyde isomers. In addition, prochiral olefins provide a racemic mixture of chiral aldehydes when achiral catalyst is used. With the employment of chiral catalysts enantioselectivity can be achieved. Starting from monosubstituted olefins the enantioselection occurs through the branched aldehyde formation (Scheme 1(b)) and enantioselectivities exceeding 90% have been obtained by using chiral rhodium and platinum complexes as catalysts. The formation of more than two aldehyde isomers may occur if hydrogen shift of the substrate takes place during hydroformylation as illustrated in Scheme 1(c).
C bonds and can be considered consequently as a very robust synthetic tool. The primary products of the exothermic reaction are aldehyde isomers. If the carbon atoms of the double bond are equivalent, only one aldehyde is formed, as depicted in Scheme 1(a). Substituted alkenes afford at least two aldehyde isomers. In addition, prochiral olefins provide a racemic mixture of chiral aldehydes when achiral catalyst is used. With the employment of chiral catalysts enantioselectivity can be achieved. Starting from monosubstituted olefins the enantioselection occurs through the branched aldehyde formation (Scheme 1(b)) and enantioselectivities exceeding 90% have been obtained by using chiral rhodium and platinum complexes as catalysts. The formation of more than two aldehyde isomers may occur if hydrogen shift of the substrate takes place during hydroformylation as illustrated in Scheme 1(c).
 | ||
| Scheme 1 Examples for hydroformylation of various olefins. | ||
Various transition metal compounds can serve as catalyst precursor for hydroformylation, such as cobalt,2 rhodium,3–6 platinum,7 ruthenium,8,9 and iridium10 complexes. Very few examples are known about the hydroformylation activity of iron complexes,11,12 although synergistic effect with rhodium complexes has been also reported.13
Hydroformylation is one of the leading industrial process among homogeneously catalysed reactions producing more than 10 million metric tons of oxo chemicals per year.14 Formed aldehydes can be valuable final products, but often they are versatile intermediates, often in tandem reactions,15 in the production of bulk chemicals, like alcohols, acids, or amines. Moreover, asymmetric hydroformylation is of great interest for the pharmaceutical and agrochemical industry, since the demand for enantiomerically pure products is steadily increasing. For instance, the hydroformylation reaction of vinyl aromatics may afford intermediates toward optically active functionalised 2-arylpropanoic acids, which serve as nonsteroidal antiinflammatory agents. For leading reviews of asymmetric hydroformylation, see ref. 16–19. The hydroformylation utilising alternative metals is reviewed recently by Beller and co-workers.20 In 2012, Franke, Selent, and Börner summarised the recent developments in the area of applied hydroformylation.14 For comprehensive reviews see the surveys of Ungváry21 and a book chapter focusing on carbonylation of alkenes and dienes.22
In the last two decades computational techniques as well as computer hardwares showed enormous progress. By now transition metal compounds can be studied by means of computational methods with high accuracy. The present review will concentrate on pivotal achievements in the field of computational chemistry related to hydroformylation discussing studies involving cobalt, rhodium, platinum and ruthenium complexes as catalysts. With the representative examples selected here the aim of this study is to give an outline how quantum chemistry has been applied to provide answers to real problems encountered in homogeneous catalysis. Transition metal computational chemistry related to homogeneous catalysis has been covered in numerous reviews.23–26 A recent review by Tsipis provides a comprehensive summary about the applicability of DFT methods in transition metal chemistry.27
2. General remarks
Recent progress in computational chemistry has shown that several important physical and chemical properties of molecules being involved in catalytic reactions can be predicted more and more accurately by employing various quantum chemical techniques, especially density functional theory (DFT) methods. In general, the more sophisticated the computational model, the more expensive in terms of computational resources. Therefore, the computational chemist needs to find the golden mean between accuracy and cost for various levels of theory.Nowadays, the vast majority of computational studies on catalytic systems involving transition metals (TMs) relies on diverse gradient-corrected DFT methods, often hybrid functionals incorporating various amount of Hartree–Fock exchange. Some examples can be found for second and forth order Møller–Plesset (MP2 and MP4)28 or CCSD(T) (coupled cluster with perturbative triples)29 calculations (latter two only for refinement of energy calculations obtained at lower level of theory) used for studies on catalytic systems. These methods are, however, somewhat less useful for first-row transition metal complexes, because 3d orbitals are much more compact than the 4s orbitals.30 The compact nature of 3d orbitals leads to the presence of strong near-degeneracy effects due to their weak overlap with ligand orbitals. The influence of near-degeneracy effects on complexes of the second- and third-row transition metals is significantly smaller, because the 4d and 5d orbitals are larger relative to the atom's size. Near-degeneracy effects often result in multireference character in the state of interest, and in principle, these states cannot be described accurately by single-reference methods such as Hartree–Fock (HF) methods and post Hartree–Fock single-determinant methods, like MP2, or coupled-cluster methods. The inappropriate description of the electronic state by Hartree–Fock, or MP2 methods may lead to erroneous geometries, and consequently, incorrect energetics. Density functional methods, on the other hand are capable to tackle the task to correctly handle complexes containing first-raw transition metals. It should be noted, however, that the lower sensitivity of DFT methods to multireference character depends on the amount of HF exchange included in the functional. Pure GGA functionals (following the generalised gradient approximations, without HF exchange) tend to be most robust, whereas the increasing amount of HF exchange may introduce instability increasingly for systems with multiconfigurational character.31
By now a vast number of DFT functionals have been developed. Among them still B3LYP is the most popular, which is the combination of Becke's three-parameter hybrid exchange functional32 with the Lee–Yang–Parr correlational functional.33 PBE0 (also known as PBE1PBE)34 is a widely used and accurate hybrid functional as well.35 The MPW1PW91 functional36 is sometimes superior in predicting geometric parameters.37
Pure GGA calculations can take advantage of the density fitting approximation.38 Instead of computing the two-electron integrals, this approach expands the density in a set of atom-centred functions when computing the Coulomb interaction, which usually results in a huge performance increase. The most frequently employed functional in this category is perhaps BP86, which is a combination of Becke's exchange functional39 with Perdew's correlation functional.40 The PBEPBE functional by Perdew, Burke, and Ernzerhof34 (also known as PBE) may serve as a quite robust selection as well. Both BP86, and PBEPBE have been reported to predict well the CO stretching frequencies in transition metal carbonyl complexes.41,42 Meta-GGA functionals such as M06-L43 or its hybrid-meta-GGA counterpart M06 (ref. 44) are promising in the prediction of molecular geometric containing transition metals.45 In a comprehensive paper by Goerigk and Grimme reported a thorough energy benchmark study of various density functionals including some recent ones with D3 dispersion correction, as well as double hybrid functionals.46
The choice of the basis set for the given calculations is usually a balancing between accuracy and computational cost. According to Xu and Truhlar the def2-TZVP47 (TZVP stands for valence triple-ζ with polarization functions) basis set is recommended for organometallic systems,48 whereas Schaefer et al. found that double-ζ basis sets often predict reliable structural parameters for transition metal complexes at a much lower cost in comparison to triple- or even quadrupole-ζ basis sets.49
It is generally accepted that the initial step of the reaction is the coordination of olefin onto the hydrido complex, which is formed under hydroformylation conditions, usually by hydrogenation of the precursor complex.50 The formation of the alkyl complex via olefin insertion is followed by the addition of CO from the carbon monoxide atmosphere and its subsequent migratory insertion into the metal–carbon bond. The oxidative addition of H2 onto the acyl complex results in a dihydrido complex from which one of the hydrido ligands transfers from metal to the acylic carbon, thus the aldehyde is released via reductive elimination and the initial hydrido complex is recovered. The generic mechanism of transition metal catalyzed hydroformylation is depicted in Scheme 2.
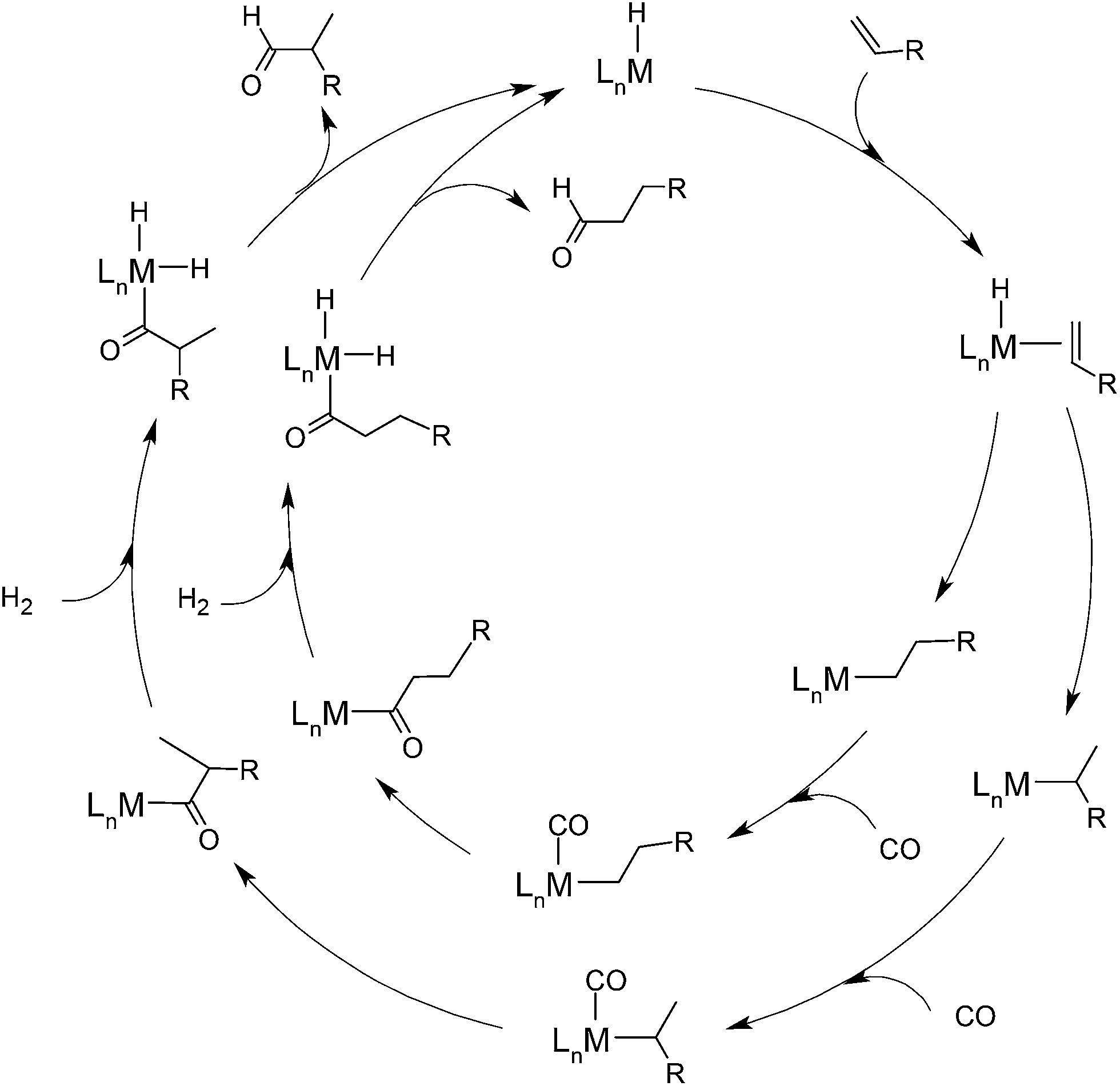 | ||
| Scheme 2 Generic hydroformylation mechanism depicting two reaction channels leading to linear and branched aldehydes. | ||
As Scheme 2 emphasizes, the bifurcation point in the mechanism is at the metal–olefin adduct, which undergoes migratory insertion affording linear and branched alkyl complexes. The leading application of hydroformylation in industry is the production of n-butyraldehyde from propene, which is further converted to 2-ethylhexanol by aldol condensation and subsequent hydrogenation. Thus, the branched isomer is undesirable in this application, however, it is the desired product in the asymmetric hydroformylation, when the goal of the reaction is one enantiomer of the branched aldehyde. Moreover, the hydrogenation of olefin may occur as side reaction, which is also an unwanted product. In all cases the control of chemo- and regioselectivity is crucial and the knowledge base leading to the design of selective catalysts is of high importance. With the aid of computational chemistry, the factors governing the selectivity may be easier to understand, and predictions can be made for the behaviour of new catalytic systems.
3. Cobalt catalysed reactions
Cobalt complexes represented the first examples for computational studies related to hydroformylation. Although performed by using the Hartree–Fock method, the studies of Antolovic and Davidson51,52 were pioneering for the understanding of behaviour of cobalt-carbonyls.3.1. Generation of the active catalyst
In 1953 Wender and co-workers reported that the oxo cobalt catalyst is the hydrido carbonyl complex HCo(CO)4 (ref. 50) whose molecular structure was described by McNeill and Scholer.53 Later, Heck and Breslow published the entire mechanism as series of equation accounting for the for the reactivity of cobalt carbonyls under oxo conditions.54 The coordinatively unsaturated 16e complex HCo(CO)3 is generated by dissociation of one of the CO ligands from the HCo(CO)4 resting state. The inhibiting effect of the CO partial pressure was investigated by kinetic studies.55 More than two decades later Wermer et al. identified the complex HCo(CO)3 with matrix infrared techniques.56 It was proved that the hydrogenation of Co2(CO)8 follows an associative pathway affording 2 molecules of HCo(CO)4 and the reaction takes place via a radical pathway involving ˙Co(CO)4.57,58 Folga and Ziegler calculated the enthalpy of hydrogenation of Co2(CO)8 with C2v symmetry59 and obtained 6.6 kcal mol−1, which is somewhat higher than the experimental 4.7 (ref. 60) and 4.1 kcal mol−1 (ref. 61) values. The homolytic dissociation of Co2(CO)8 to two molecules of ˙Co(CO)4 was investigated by Barckholtz and Bursten62 and obtained a notably higher value for the Co–Co bond dissociation enthalpy (29.9 kcal mol−1) than the experimental BDE (19 ± 2 kcal mol−1) reported by Klingler and Rathke.63,64The electronic and molecular structure of the coordinatively saturated and unsaturated hydrido cobalt carbonyls were investigated by Ziegler and co-workers at the BP86 level of theory in combination with an STO triple-ζ basis set.65 In the global minimum of singlet HCo(CO)4 the hydride ligand was found to take the axial position with a fairly strong energetic preference of 13.9 kcal mol−1 over the isomer with an equatorial hydride. Fig. 1 depicts a diagram in which the upper valence orbitals of the trigonal bipyramidal HCo(CO)4 and those of the two possible structures of HCo(CO)3 are correlated for the singlet states. The dissociation of one carbonyl ligand from HCo(CO)4 with an electronic configuration 1A1[(1e)4(2e)4] affords two unsaturated structures. The complex with Cs symmetry, with the electronic configuration 1A1[(1a′)2(2a′)2(1a′′)2(2a′′)2] proved to be more stable by 8.4 kcal mol−1 than the C3v structure. The dissociation energy of a CO ligand in HCo(CO)4 was reported as much as 49.5 kcal mol−1 for the axial CO, and 40.5 kcal mol−1 for the equatorial ligand. This gives a good explanation why high temperature is required to generate a sufficient concentration of the catalytically active species HCo(CO)3.
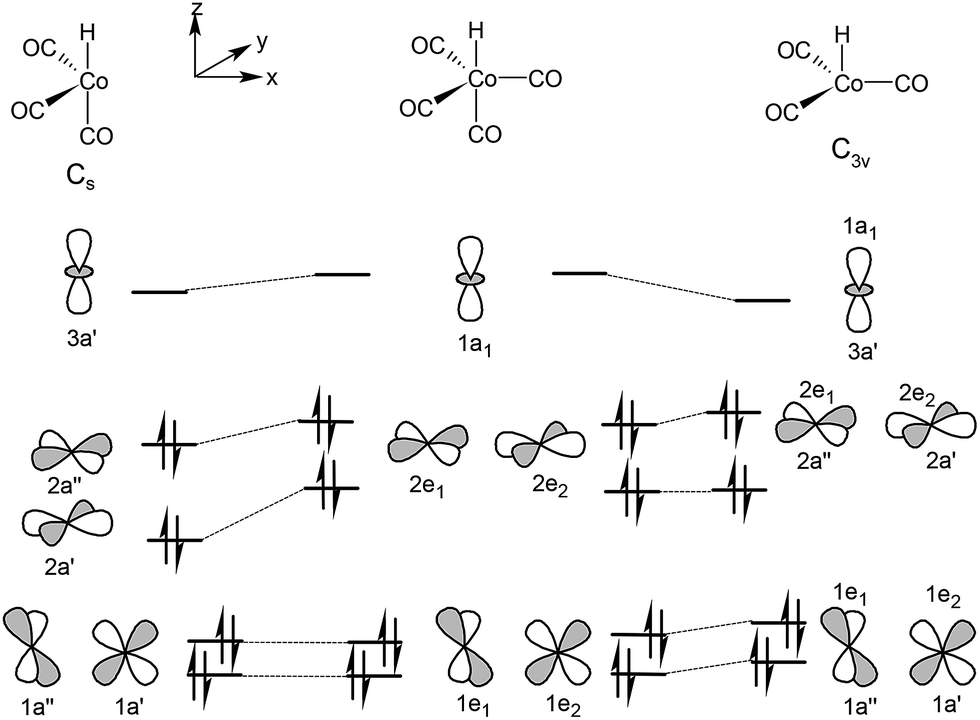 | ||
| Fig. 1 Correlating orbitals of HCo(CO)4 with orbitals of HCo(CO)3 in the two singlet states with Cs and C3v symmetry. | ||
The study on cobalt carbonyls and cobalt carbonyl hydrides by Beller et al. provided somewhat different result for the structure of HCo(CO)3 and for the energetics of its formation at the B3LYP/6-311+G(d) level of theory. The bond dissociation energy for the equatorial CO in HCo(CO)4 was 25.7 kcal mol−1, whereas the BDE for the axial CO was 36.3 kcal mol−1. For the Co–H homolysis 54.9 kcal mol−1 was calculated, thus the CO dissociation is more favoured energetically, then the homolytic cleavage of the cobalt–hydride bond. The equilibrium structure for the more stable isomer of HCo(CO)3 adopts a planar structure with C2v symmetry, in contrast with the finding of Ziegler et al. The computed geometry of HCo(CO)3 with two different levels of theory is compared in Fig. 2.
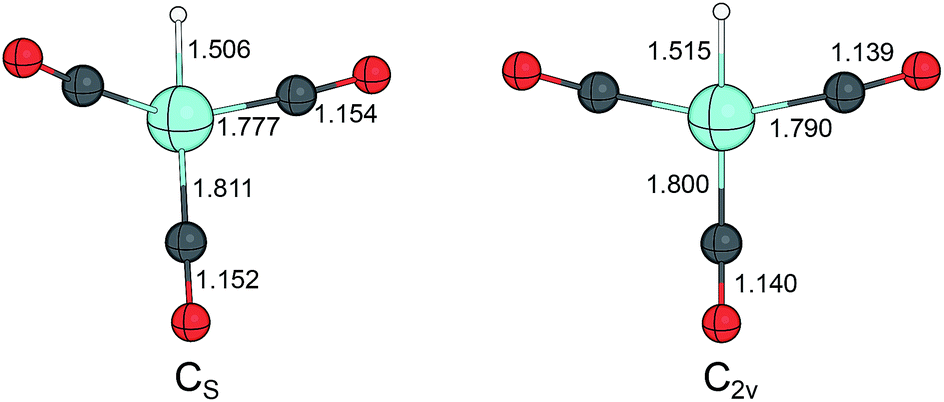 | ||
| Fig. 2 Computed structure of HCo(CO)3 at BP86 (ref. 65) and B3LYP66 with Cs and C2v symmetry, respectively. Bond distances are given in Å. | ||
3.2. Mechanism of hydroformylation with various substrates
Because of the complexity of the entire reaction mechanism, most computational works has focused on particular steps of the catalytic cycle. In 1987 Antolovic and Davidson reported the structure of HCo(CO3) (ethylene) and HCo(CO3)(ethyl) complexes computed at the HF level. The energy values were further refined by configuration-interaction calculations. For the ethylene insertion a barrier of 17.4 kcal mol−1 was obtained.52The migratory insertion of the carbonyl group into the Co–Cmethyl bond was studied by Goh and Marynick at B3LYP level.67 It was found that one of the Co(CO3)(COCH3) acyl intermediates was stabilised by the acyl oxygen adopting an η2 type of coordination. The energy barrier for the combined step (CO insertion and subsequent rotation of the Co–acyl bond) was 15.9 kcal mol−1. The formation of the acetyl complex was found to be slightly endothermic by 2.7 kcal mol−1.
The entire catalytic cycle of the hydroformylation of propene catalysed by HCo(CO)3 was calculated by Huo, Li, Beller, and Jiao (HLBJ).68 They found that the insertion of the olefin into the Co–H bond takes place through a migratory insertion transition state, accompanied by a simultaneous rotation of the Co(CO)3 group, leading to the stable alkyl complex with the propyl groups at the axial position both for the linear and branched pathways, and stabilised by agostic interaction at the formally vacant equatorial position. The alkylation step was found to be an equilibrium process, thus reversible. The subsequent CO addition to the alkyl complexes, however, was found to be exothermic. It was concluded, that the corresponding linear and branched complexes RCo(CO)4 should be responsible for the regioselectivity, which is a thermodynamically controlled process. The computations reveal that the formation of the linear product is more stable by 2.1 kcal mol−1, hence more favoured than the branched one, in line with the experimental observations. The Co(CO)4(propyl) key intermediates are depicted in Fig. 3.
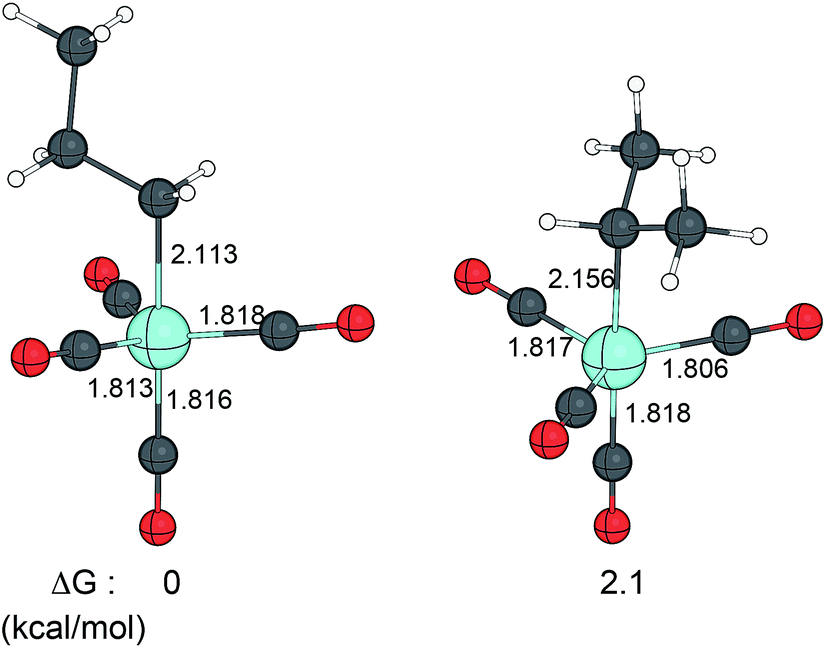 | ||
| Fig. 3 Linear and branched Co(CO)4(propyl) complexes. Bond distances are given in Å. | ||
The mechanism of the cobalt catalysed hydroformylation of acetylene was also investigated by HLBJ.69 Acetylene hydroformylation was revealed to be more favoured energetically than the competitive acetylene hydrogenation, therefore the formation of the saturated aldehyde takes place via subsequent hydrogenation of the initially formed α,β-unsaturated aldehyde rather than via hydrogenation of acetylene prior to hydroformylation. In contrast to propene, acetylene insertion into the Co–H bond is a fast and irreversible process, which is assumed to determine the regioselectivity of the reaction of terminal alkynes. The free energy barrier is 6.3 kcal mol−1, and the step is exergonic by −20.8 kcal mol−1 (Fig. 4).
 | ||
| Fig. 4 Free energy profile for the acetylene coordination and insertion into the Co–H bond. | ||
The carbonylation of the vinyl complex results in the relatively stable η3-acyl complex, which is more stable by 3.2 kcal mol−1 than the acyl complex stabilised by an η3 coordination of the acyl oxygen (Fig. 5).
 | ||
| Fig. 5 Cobalt carbonyl α,β-unsaturated acyl complexes with η3 and η2 coordination. Bond distances are given in Å. | ||
The next stage in the series of systematic studies of cobalt catalysed reactions by HLBJ was the investigation of the hydroformylation of butadiene.70 Three possible pathways leading to unsaturated monoaldehydes were proposed, as depicted in Scheme 3. The anti-Markovnikov reaction, with the formation of linear aldehyde in 1,2-addition leading to 4-pentenal, was found to be reversible, whereas the pathway associated with Markovnikov insertion is irreversible due to the enhanced stability of the syn-η3-allyl complex.
 | ||
| Scheme 3 Proposed mechanism for butadiene hydroformylation resulting in unsaturated aldehydes. | ||
The CO addition to the syn-η3-allyl species favours the eventual formation the 1,4-addition product over the branched 1,2-addition aldehyde, both kinetically and thermodynamically.
The hydroformylation of alkynes and allenes is a potential synthetic route for preparing α,β-unsaturated aldehydes, which are valuable intermediates in fine chemicals and pharmacy. This type of reaction was studied by HLBJ employing B3LYP/6-311+G(d) level of theory.71 The relative stability of the syn-η3-allylic intermediate was found to account for the regioselectivity as the linear anti-Markovnikov intermediate was more stable by 16.3 kcal mol−1 than the branched Markovnikov intermediate (Fig. 6). It was concluded that propyne hydroformylation is not regioselective since the formation of the linear anti-Markovnikov and branched Markovnikov intermediates had equal probability. The coordination mode of allene was also studied and only the equatorial structure was determined as a genuine minimum, whereas the syn and anti structures (with respect to the CH2 group) were identified as rotational transition states (Scheme 4).
 | ||
| Fig. 6 Hydroformylation of allene and propyne (top); key intermediate structures for allene insertion (bottom). Bond distances are given in Å. | ||
 | ||
| Scheme 4 Computed structure of the initial allene complex (a); and transition states with syn (b) and anti (c) arrangement of the CH2 group. | ||
Rush, Pringle, and Harvey built a kinetic model for the phosphine-free cobalt-catalysed hydroformylation employing the CCSD(T)-F12 protocol and transition state theory.72 At this level 25.9 kcal mol−1 was calculated as the enthalpy for the hydroformylation of propene, which is in very good agreement with the room temperature experimental value of 26.3 kcal mol−1.
They found that the coordination of alkene or CO proceeds without barrier, so the corresponding transition states are purely entropic in origin. These steps were assumed to be diffusion-controlled, with rate constants in solution given by the approximate expression k = 8kBT/3η, where kB is Boltzmann's constant, T is temperature, and η is the solvent viscosity.73
The authors assumed that the processes with low activation barriers are in quasi-equilibrium throughout, and the reductive elimination steps leading to aldehyde (or alkane, in the hydrogenation side reaction) are irreversible. Thus, only the forward rate constants needed to be included for kh and kp. The reduced mechanism is depicted in Scheme 5.
 | ||
| Scheme 5 Reduced mechanism of cobalt-catalysed hydroformylation of propene. | ||
The steady-state approximation was applied to the concentration of all cobalt-complexes. The concentration of other species were assumed to be unchanged. The set of steady-state kinetic equations yielded the expression (eqn (1)) for the rate of the catalytic reaction R as a function of the concentrations of HCo(CO)4 (1), propene, the partial pressures of hydrogen (pH2) and carbon monoxide (pCO).
 | (1) |
The rate constants are shown in Scheme 5, whereas KD and K1 (=k1/k−1) are equilibrium constants. By using eqn (1) the rate for propene hydroformylation was computed for various set of concentrations and excellent agreement with the experimental rates were obtained.
The artificial force induced reaction (AFIR) method,74 developed by Maeda and Morokuma was applied to explore the first step, that is the olefin insertion, of cobalt-catalysed hydroformylation of propene.75 The AFIR method was originally developed for intermolecular reactions for finding associative reaction pathways between two or more molecules, but can be applied also for intramolecular reactions by manually dividing the molecule into fragments. This approximation is denoted as SC-AFIR. All reactive sites among multiple reactants can be identified as local minima on an AFIR function. By locating reactive sites an additional force causes geometrical changes that are similar to those in a reaction pathway and provides approximate intermediate (local minima on the PES) and transition state structures. These approximate structures are then re-optimised to obtain the true transition state and product structures. For the olefin insertion step all the possible pathways were correctly identified by employing the SC-AFIR method.
The AFIR method was also employed for the semiautomatic determination of the entire catalytic cycle of ethylene hydroformylation catalysed by the unmodified cobalt catalyst without using any initial guess.76 The AFIR method was performed at the relatively cheap B3LYP/6-31G level to save computer time. All the products, intermediates and transition states were reoptimised at the M06/6-311++G(d,p) level with the zero-point energy and free-energy corrections in an experimental condition (403.15 K, 200 bar). Results at the M06 level were qualitatively similar to those obtained at B3LYP level. The catalytic cycle resulting in acetaldehyde as product agrees well with the Heck–Breslow mechanism.
4. Rhodium catalysed hydroformylation
The assumed mechanism for rhodium-catalysed hydroformylation with the unmodified catalyst HRh(CO)4 corresponds to the Heck–Breslow mechanism54 of the cobalt-catalysed reaction. For this system the catalyst precursor is tetrarhodium dodecacarbonyl which fragments to mononuclear species under hydrogen pressure.77The mechanisms for the unmodified and ligand-modified system are assumed to be almost identical. For systems modified by various P-donor ligands precursors like [Rh(CO)2(acac)] (acac = acetylacetonate) or [Rh(COD)(acac)] (COD = 1,5-cyclooctadiene) are employed.3 In 1965 Wilkinson et al. reported the hydroformylation of 1-hexene employing the [RhCl(PPh3)3] precursor.78 The reaction conditions (55 °C and 90 bar CO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2 = 1:1) were unusually mild in comparison to the earlier cobalt containing systems. The reaction proceeded also with superior chemo- and regioselectity.79–81 Subsequently, rhodium-based catalytic systems gained considerable attention and huge progress in terms of chemo- and regioselectity has been made to date. The Ruhrchemie/Rhône-Poulenc process utilises a water-soluble rhodium catalyst in a two-phase hydroformylation process.82,83 Also hydroformylation is one of the most powerful methods for the functionalization of carbon–carbon double bond hence a very effective tool for the preparation of fine chemicals.15 Optically active aldehydes arising from asymmetric hydroformylation are of great interest because they may serve as synthetic intermediates for the production of sophisticated pharmacologically active molecules. The aldehyde group is one of the most versatile functional groups, thus a variety of useful chiral chemicals such as alcohols, acids and amines can be easily prepared from chiral aldehydes.
:H2 = 1:1) were unusually mild in comparison to the earlier cobalt containing systems. The reaction proceeded also with superior chemo- and regioselectity.79–81 Subsequently, rhodium-based catalytic systems gained considerable attention and huge progress in terms of chemo- and regioselectity has been made to date. The Ruhrchemie/Rhône-Poulenc process utilises a water-soluble rhodium catalyst in a two-phase hydroformylation process.82,83 Also hydroformylation is one of the most powerful methods for the functionalization of carbon–carbon double bond hence a very effective tool for the preparation of fine chemicals.15 Optically active aldehydes arising from asymmetric hydroformylation are of great interest because they may serve as synthetic intermediates for the production of sophisticated pharmacologically active molecules. The aldehyde group is one of the most versatile functional groups, thus a variety of useful chiral chemicals such as alcohols, acids and amines can be easily prepared from chiral aldehydes.
4.1. Generation of the active catalysts
The equilibrium structures of rhodium complexes HRh(CO)n(PH3)4−n and HRh(CO)n(PH3)3−n (n = 1–3) were investigated computationally84 using the local density approximation (LDA) for geometry optimisations at MP2 and CCSD(T) levels for calculating the dissociation energies. In these systems MP2 strongly overestimated the bond strengths in comparison to the coupled cluster calculations. On the other hand, BP86 calculations yielded energy values close to the CCSD(T) energies.The coordinatively unsaturated complexes trans-HRh(CO)2(PH3) and HRh(CO)3 adopt a nonplanar structure with a bond angle of approximately 150° between the carbonyl ligands in trans position. These two complexes have been recomputed with the dispersion-corrected GGA functional B97-D3 (ref. 85) in combination with the def2-TZVP basis set47 and the obtained geometries showed very close resemblance to those calculated previously at the LDA level (Fig. 7). The dissociation energies using PH3 as model ligands were too low in comparison to the experimental values for PPh3 complexes.86 Employing PMe3 as ligand, the phosphine and carbonyl dissociation energies become much closer, thus the substitution of a phosphine by CO does not continuously stabilise the complexes. Thus, both HRh(CO)2(phosphine) and HRh(CO)(phosphine)2 complexes can be candidates for olefin coordination under usual reaction conditions.
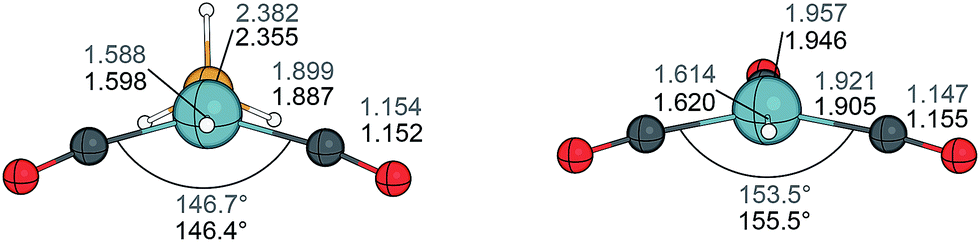 | ||
| Fig. 7 Optimised geometries of the four-coordinate complexes HRh(CO)2(PH3) (left) and HRh(CO)3 (right) computed at the LDA level (ref. 84, black) and the B97-D3 level (this work, grey). Bond distances are given in Å. | ||
The ligand dissociation reactions of complexes HRh(CO)2(PR3)2 (R = H, F, Me) were investigated by Gleich and Hutter.87 In these trigonal bipyramidal complexes the phosphine ligand can be arranged in axial-equatorial or diequatorial manner. For PH3 the diequatorial complex is almost in equilibrium with the axial-equatorial species, whereas for the PF3 case it is more stable by 2.3 kcal mol−1. On the other hand, for the more basic PMe3 the axial-equatorial arrangement of phosphines is preferred. Based on HF88 and MP2/HF calculations89 Morokuma and co-workers also assumed that the four-coordinate phosphine–dicarbonyl complex reacts with olefin in the alkyl formation step. It was also concluded that solvent effects are not negligible even when hydrocarbon solvents are considered because the coordinatively unsaturated four-coordinate intermediates undergo a significant stabilisation upon solvation in ethylene.
The CO dissociation from HRh(CO)4 resulting in HRh(CO)3 was found to be endothermic by 27.7 kcal mol−1 according to MP2 calculations. At the BP86/TZVP level 21.1 kcal mol−1 was obtained which is very close to the value calculated at the CCSD(T)/TZVP//BP86/TZVP level (20.7 kcal mol−1). In the phosphine-carbonyl complexes the dissociation of phosphine is preferred over that of CO regardless of the basicity of phosphine (Fig. 8).
 | ||
| Fig. 8 Ligand dissociation reactions for complexes HRh(PR3)2(CO)2. Energies were computed at the BP86/TZVP level. | ||
Decker and Cundari examined the formation of bis phosphine complexes HRh(PR3)2(CO) at the B3LYP and CCSD(T)//B3LYP levels of theory.90 Phosphine dissociation was found to be endothermic by 13.8 and 15.4 kcal mol−1 (at the CCSD(T)//B3LYP level) leading to trans- and cis-HRh(PR3)2(CO), respectively. The preferred pathway for the catalytic cycle originates from the trans isomer.
Grigoropoulos and co-workers synthesised chalcogenidoimidodiphosphinato–Rh complexes and employed them in the hydroformylation of styrene.91 The structure of the catalyst precursors were computed at BP86/TZVP level (Fig. 9). In the dicarbonyl complex one CO can be easily replaced by PPh3 which is, however, significantly decreases the catalytic activity of these systems.
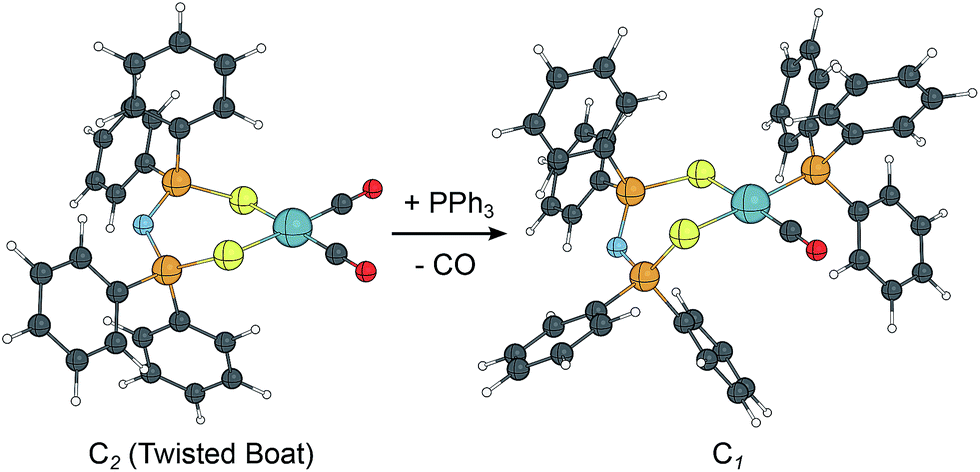 | ||
| Fig. 9 Computed structures of Rh–chalcogenidoimido–diphosphinato-carbonyl complexes at the BP86/TZVP level of theory. | ||
The catalytically active complex HRh(CO)2L with a triptycene-derived bisphosphine ligand was studied in solution, in the solid state, and computationally in order to determine the molecular structure and to interpret the equilibria between isomers.92 The predicted intramolecular distance of the two biphenyl side groups of these ligands strongly depends upon the treatment of inter- and intramolecular noncovalent interactions. Low-temperature NMR studies confirmed the equilibrium between the axial-aquatorial and diequatorial isomers. To reproduce well the experimental findings the use of solvation corrections even for geometry optimisations was important, especially for DFT-D calculations.
4.2. Mechanism of rhodium-catalysed hydroformylation with various substrates
The potential energy hypersurface for ethylene hydroformylation was scrutinised at the B3LYP level in combination of the SBK basis set,93 and the energies were also evaluated at the CCSD(T) level.90 Both cis- and trans-HRh(PH3)2(CO) were considered as active catalysts. The olefin insertion is predicted to be slightly exothermic for both pathways with a kinetic preference for the axial-equatorial arrangement of the phosphine ligands. The exothermic CO coordination to the ethyl complexes is followed by the relatively slow and endothermic insertion step resulting in the propionyl complexes. The oxidative addition of H2 is exothermic, with barriers less than 10 kcal mol−1. The fairly slow and irreversible elimination step results in propionaldehyde regenerating the four coordinate hydrido rhodium carbonyl catalysts. The transition states for the olefin and CO insertion steps are depicted in Fig. 10. | ||
| Fig. 10 Computed structure of transition states involving in the ethylene insertion and CO insertion steps. Bond lengths are given in Å, relative energies in kcal mol−1. Energy values given in bold correspond to CCSD(T)//B3LYP relative energies, while those in parentheses correspond to B3LYP//B3LYP relative energies. | ||
The energy of hydroformylation (that is the energy difference between the aldehyde product and the reactant components: ethylene, CO, and H2) is predicted to be −34.7 kcal mol−1 at the B3LYP level. Refinement of energies at the CCSD(T) level yielded a value of −24.4 kcal mol−1.
Gleich and Hutter studied the catalytic cycle of ethylene hydroformylation with unmodified hydrido-Rh(I) carbonyl system, as well as with modified systems containing phosphine ligands with different basicity.87 Apart from static calculations at the CCSD(T)/TZVP//BP86/TZVP level of theory, CPMD dynamic calculations94 were also performed. The high activity of the unmodified system was explained by the unhindered olefin association, while phosphine containing systems suffer from association barriers depending on their steric demands. Moreover, the olefin association becomes thermodynamically more favoured with decreasing basicity. The greater sensitivity of the unmodified catalyst towards H2 pressure can be explained by a less favourable energy profile of the hydrogen addition.
QM/MM study within the ONIOM framework95 aimed at the determining how phosphine substituents influence the energetics of the insertion of ethylene into the Rh–H bond.96 Geometry optimisations for PtBu3 resulted in dissociation of one of the phosphine ligands, suggesting that HRh(PtBu3)(CO)2 is the active species, which is sterically less crowded in comparison to the hydrido bis-phosphino monocarbonyl species. In the case of arylphosphines, as spectator ligands, a clear kinetic as well as thermodynamic energetic preference was found for ethylene insertion to proceed from the least stable diequatorial ethylene adduct to the most stable cis Rh-ethyl insertion product via a diequatorial transition state. For trimethylphosphine both the diequatorial and the axial-equatorial pathways seem feasible.
The origin of stereodifferentiation in rhodium-catalysed hydroformylation was elucidated by the example of the bidentate phosphine–phosphite ligand BINAPHOS (Scheme 6) employing a semiquantitative theoretical model.97 It was demonstrated that the outstanding properties of BINAPHOS are due to the combination of three factors: pronounced coordination preferences for steric and electronic reasons; adequate number of chirality elements; (iii) correct configuration of the binaphthyl fragments.
 | ||
| Scheme 6 BINAPHOS, and bidentate ligands possessing C2 symmetry. | ||
A combined QM/MM method with frozen reaction centres was applied for connecting the phosphine coordination modes to the regioselectivities.98 Tendencies in regioselectivities of systems with chelating ligands DIPHOS (also known as DPPE), BISBI, and NAPHOS (Scheme 6) and triphenylphosphine as reference were reproduced. Irreversible olefin insertion and no changes in the n/iso distribution after the formation of Rh-alkyl intermediates was assumed and the formula given in eqn (2) was employed:
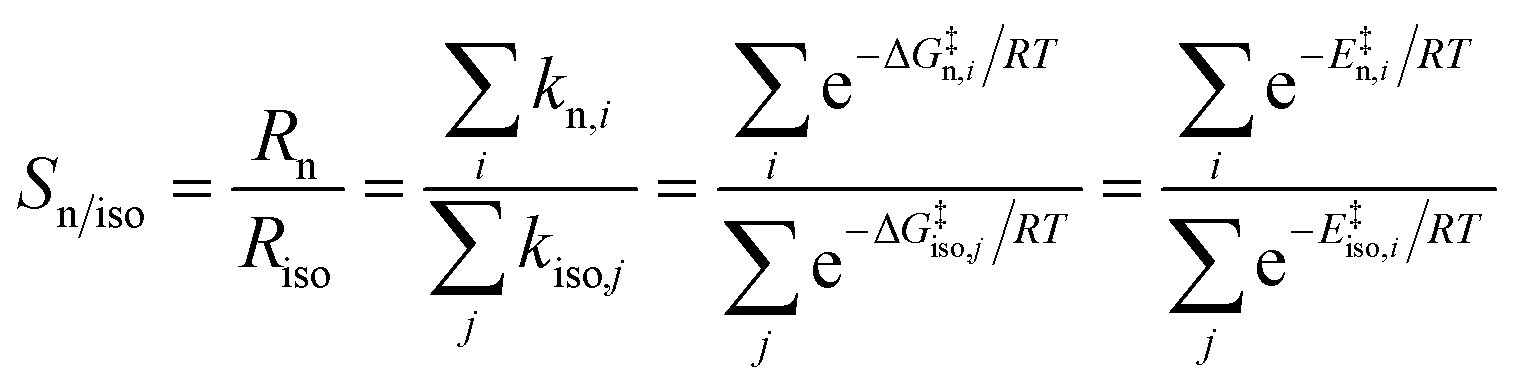 | (2) |
The stereoselectivities of Rh-containing systems containing C2-symmetric ligands CHIRAPHOS, BINAP, DIOP, and NAPHOS (Scheme 6) were calculated with a combined QM/MM method.99 It was assumed, that all ligand coordination modes favour transition states with the same asymmetric induction. The performance of C2-symmetric bidentate phosphine ligands was concluded to be governed by two interdependencies, namely the induction influence of the chelate ring and the flexibility of the ligand backbone. The smaller the chelate ring (like in the case of CHIRAPHOS), the more diminished its influence on asymmetric induction. A larger chelate ring often results in a greater backbone flexibility, and the diequatorial coordination mode must be additionally considered, which can impair the total selectivity. The success of some C1-symmetric ligands, like BINAPHOS may be due to the reduced number of coordination modes, and the right number and configuration of chirality centres.
The insertion of propene into the H–Rh bond starting from HRh(PH3)2(CO)(propene) was investigated by Rocha and de Almeida at the MP4(SDQ)//BP86 level using double-ζ basis sets.100 The diequatorial olefin adduct was considered as initial compound. The formation of four-coordinate alkyl complexes Rh(PH3)2(CO)(propyl) was found to be reversible. Comparing the two concurrent transition states the branched pathway is predicted to be faster by 1.9 kcal mol−1. The branched alkyl complex, however, is less stable by 2.1 kcal mol−1 than the linear isomer, and using Boltzmann distribution, the ratio of the normal and iso products is 96:4, which is in agreement with the experimental regioselectity 95:5 obtained by water soluble catalyst.83
The olefin insertion and carbonylation steps for the Rh-catalysed hydroformylation of styrene was studied at the MP4(SDQ)//BP86 level using the model catalysts of the type [HRh(CO)x(PMe3)3−x].101 It was proposed, that under normal hydroformylation conditions the active catalytic species is trans-HRh(CO)(PMe3)2. The coordination energy of styrene to the rhodium centre is quite sensitive to the theoretical method employed as the MPn calculations were found to overestimate the stability of the π-complexes in comparison to the BP86 values. Despite these differences, the absolute values of activation and reaction energies between the distinct pathways, leading to branched and linear species, are almost the same. The CO coordination followed by insertion into the metal–Calkyl bond reveals different patterns for the monocarbonyl and dicarbonyl catalysts. In the presence of trans-HRh(CO)(PMe3)2 the linear Rh-acyl intermediate is preferred kinetically, but the branched intermediate is preferred thermodynamically. With complex HRh(CO)2(PMe3), however, the branched Rh-acyl intermediate is not favoured neither kinetically, nor thermodynamically.
The electronic and steric effects of various phosphine ligands on the selectivity of the propene and styrene insertion into the Rh–H bond of the complexes HRh(CO)2(PR3)(olefin) (where R = H, F, Et, Ph, OEt, and OPh) is investigated at the M06 level of theory with triple-ζ basis sets.102 Good correlation was found between the Tolman electronic factor χ (ref. 103) and the backdonated charges from the metallic centre to the olefin as well as with the interaction energy of the olefin. When the substrate is propene, the pathway leading to the linear metal-alkyl intermediate is preferred over the branched route for all phosphines and phosphites considered. According to the authors, however, it cannot be clearly concluded that the regioselectivity of the reaction is resulted from kinetic or thermodynamic control. For styrene, the branched metal-alkyl intermediate is strongly favoured in all cases. Moreover, the activation energy for all P-donor ligands is smaller for the pathway leading to the branched-alkyl species.
The hydroformylation of ethylene for the monosubstituted Rh-carbonyl precursor HRh(CO)3L (where L = P(OMe)3, P(OPh)3, P(OCH2CF3)3, PMe3, PEt3, PiPR3, PPh3, or a N-heterocyclic carbene) was scrutinised by Jensen and co-workers employing the OLYP functional33,104 in combination with double-ζ basis sets.105 For the catalysts containing electron-donating ligands the rate-determining step is the coordination-insertion of ethylene. For electron withdrawing phosphites, and for the good π acceptor NHC ligands 18-electron acyl rhodium intermediates were identified as new resting states, and the rate-determining step was found to be the reductive elimination of the aldehyde. The barrier for the ethylene insertion revealed strong dependence on the electronic properties of the spectator ligands. For instance, almost twice as much barrier was predicted for the NHC ligand N,N-dimethyl-hexahydropyrimidine in comparison to that calculated for triphenylphosphite (Fig. 11). The rate-controlling barriers are increased in the ordering of ligands CO < strongly electron withdrawing phosphites < standard phosphites < aryl phosphines and PMe3 < higher alkyl phosphines in qualitative agreement with experimental observations.
 | ||
| Fig. 11 Computed structures of the catalytically active complexes of HRh(CO)3L type (where L = P(OPh)3, or the NHC N,N-dimethyl-hexahydropyrimidine) and the transition states describing the ethylene insertion into the Rh–H bond. Bond distances are given in Å. | ||
Alagona and co-workers used the unmodified rhodium catalyst HRh(CO)3 for their theoretical investigation on hydroformylation of a large set of substrates, such as propene (a), 2-methylpropene (b), 1-hexene (c), 3,3-dimethylbutene (d), fluoroethene (e), 3,3,3-trifluoropropene (f), styrene (g), vinyl-methylether (h), and allylmethylether (i).106 The B3P86 hybrid functional was employed in combination with double-ζ basis sets. The olefin insertion was studied as the crucial step, determining the regioselectity of the whole process. Fig. 12 depicts the branched and linear pathways for the insertion of styrene into the Rh–H bond. The regioselectity was estimated making use of the formula as given in eqn (2) and, for comparison, it was also evaluated experimentally for most of the substrates. The agreement between computed and experimental data was very good in general, although selectivities close to 50% are more difficult to estimate accurately, because they are much more sensitive to small energy differences, than those close to 100% for either of the isomers. According to the authors the internal energy was preferable to the use of free energy when the substrates are prone to internal rotations, which is especially the case for propene and 1-hexene, which in the harmonical vibrational analysis are considered as true vibrations, thereby affecting the thermodynamic functions.
 | ||
| Fig. 12 Structures and energies along the branched and linear pathways for the rhodium-catalysed hydroformylation of styrene. | ||
Both steric and electronic effects determine the n/iso ratio when the Rh-alkyl formation is the regioselectivity determining step. As the carbon in α position, with respect to the Rh centre, carries some negative partial charge, π-acceptor ligands, like aryl, or alkoxy stabilise the charge distribution to a greater extent for the branched alkyl intermediate (Scheme 7). On the other hand, in the presence of the electron donating alkyl substituents the formation of the linear alkyl-complex is preferred energetically.107
 | ||
| Scheme 7 Stabilization of alkyl-rhodium intermediates arising from the hydroformylation of different alkenes possessing electron withdrawing or electron donating groups. | ||
Deuteroformylation experiments carried out at partial substrate conversion is an appropriate tool to determine whether the olefin insertion step is reversible or not.108–112 For substrates like 1-hexene, styrene and allylethylether110 no deuterated olefin is detected when the reaction was performed under mild conditions and hence the isomeric metal-alkyl intermediate are directly transformed into the acyl species to give rise to isomeric aldehydes preserving the normal/iso ratio. The 2H spectrum of a mixture resulting from deuteroformylation of ethylvinylether at 100 °C showed the presence of Et–O–CDCH2 and Et–O–CHCHD indicating that β-hydride elimination occurs at higher temperature. In the case of styrene the β-hydride elimination takes place only for the branched intermediate when the temperature is lower than 100 °C.
The hydroformylation of 1,1-diphenylethene reveals, however, a remarkable difference for the linear and branched pathways. While the former one, similarly to that of styrene or 1-hexene, consists of an irreversible olefin insertion step, for the branched alkyl complex the β-hydride elimination is significantly slower, then the CO insertion step. Hence, the branched conformers leave their pathway and return to reactants resulting in eventually an enrichment in the linear product.113
The comparison of branched and linear pathways for the Rh-catalyzed hydroformylation of the bulky chiral substrate 3,4,4-trimethylpent-1-ene computed at B3P86 level shows a distinct contrast as well.114 The alkyl formation step is irreversible for all linear isomers that yield the linear aldehyde. Some branched Rh-alkyl isomers eventually afford the corresponding aldehydes as well, two others follow competing pathways due to the low relative barrier of β-hydride elimination. When the barrier for CO addition and insertion is high, the Rh–olefin complex forming from the Rh-alkyl complex can follow the linear pathway in a concurrent reaction. Moreover, internal olefin–Rh complexes can form as well, when the CO insertion step is slow enough.
The insertion of chiral olefins, such as (1-vinyloxy-ethyl)-benzene, (1-methyl-but-3-enyl)-benzene, and (1-methyl-allyl)-benzene into the H–Rh bond of unmodified catalyst HRh(CO)3 was investigated at the B3P86 level of theory with basis sets of double-ζ quality.115 The appearance of the second chiral centre at the inner olefin carbon upon complexation do not show significant basis set dependence. A qualitative agreement with the available experimental regio- and diastereoselectivities were obtained for the (1-vinyloxy-ethyl)-benzene and (1-methyl-but-3-enyl)-benzene substrates. For the hydroformylation of the ethereal substrate a branched regioselectity of 72% and a diasteroselectivity of 97% (for the branched diastereomers) were computed which compared well with the experimental results 85% and 88%, respectively.116 For (1-methyl-but-3-enyl)-benzene the lack of chiral discrimination was explained by the conformational flexibility of the carbon–carbon bond adjacent to the vinyl group.
The regioselectity of the hydroformylation of 2-methyl-3-(3-acetylpyrrol-1-yl)prop-1-ene catalysed by HRh(CO)3 was studied at the B3LYP/6-31G* level (LANL2DZ for Rh).117 A linear regioselectivity of 88% was obtained, in satisfactory agreement with experiment producing only the chiral linear aldehyde, which undergoes a complete diastereoselective cyclization yielding the 1:1 mixture of 1-acetyl-6R(S)-methyl-8R(S)-hydroxy-5,6,7,8-tetrahydroindolizine (having the same configuration on both stereogenic carbon atoms) and 2-acetyl-6-methyl-5,6-dihydroindolizine118 (see Scheme 8).
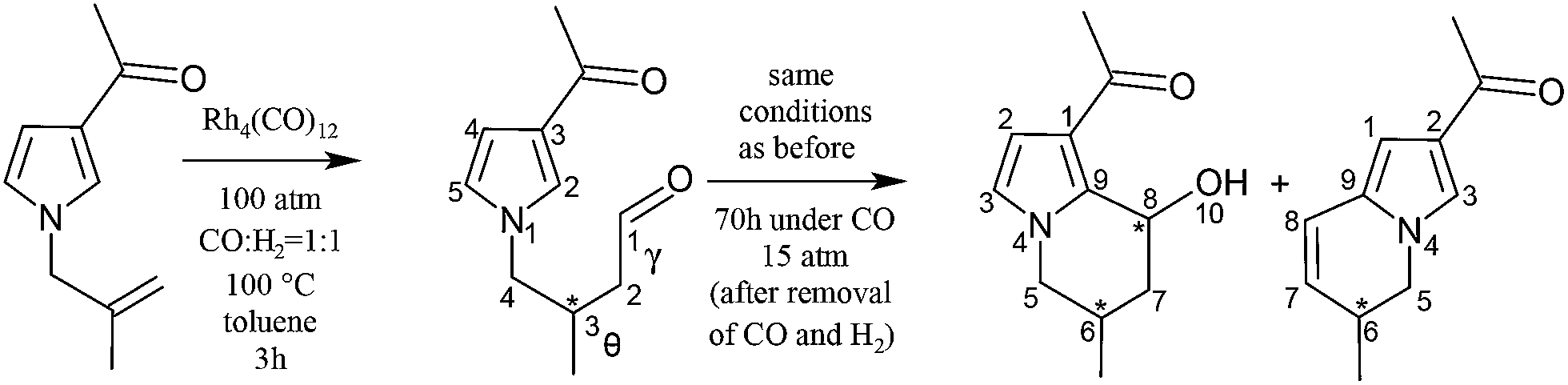 | ||
| Scheme 8 Hydroformylation of 2-methyl-3-(3-acetylpyrrol-1-yl) prop-1-ene followed by the reaction of a producing a 1:1 mixture of 1-acetyl-6-methyl-8-hydroxy-5,6,7,8-tetrahydroindolizine and 2-acetyl-6-methyl-5,6-dihydroindolizine. | ||
The hydroformylation of various olefins and acetylenes was investigated by Luo and co-workers employing the B3LYP functional in combination with double-ζ basis sets. For the elucidation of the mechanism of the reaction of 4-pyridylethene HRh(CO)(PH3)2 was considered as active catalyst. The calculations showed that the rate-limiting step is the H2 oxidative addition, and the regioselectity is originated from the olefin insertion into the Rh–H bond.119 The same elementary step was reported as rate determining step for the asymmetric hydroformylation of vinyl formate as well. The reaction was modelled with a CHIRAPHOS-type ligand with phenyl groups replaced by methyls on the phosphorous atoms. The formation of Rh-alkyl intermediates was found as selectivity determining step and the branched (S)-1-formylethyl formate was predicted as main product.120 For the competitive hydroformylation and hydrogenation of acrylaldehyde the hydrogen addition was proposed to be the rate-limiting step as well. Interestingly, for the branched intermediates the hydrogenation, whereas for the linear intermediates the hydroformylation is preferred kinetically. Moreover, the acrylaldehyde insertion for the linear alkyl complexes is revealed to be reversible, but the insertions for the branched intermediates were irreversible, thus, the regioselectivity is controlled by thermodynamics.121
The regioselectity for the hydroformylation of ethyne122 and propyne123 by the model catalyst HRh(CO)2(PH3) was found to be determined by the irreversible acetyenyl insertion step into the H–Rh bond. For ethyne this is also the rate-limiting step, whereas for propyne the rate is determined by the H2 addition. Remarkable difference between the two substrate is that the barrier for the hydrogenation of ethyne is lower than that for hydroformylation so hydrogenation is the preferred reaction in this case. The opposite trend is predicted for propyne, where the hydroformylation is predicted to take place with a lower barrier and the main product is trans-butenal. The difference in chemoselectivity for the two substrates is explained by the electronic effect of the methyl group which is present is propyne.
The origin of regioselectity in Rh-diphosphine catalysed hydroformylation was studied by means of QM/MM calculations by Carbó and co-workers.124 The IMOMM method method125 was used with B3LYP functional for the quantum mechanics part, and with the MM3 force field126 for the molecular mechanics part. The role of the diphosphine bite angle and of the nonbonding interactions were analysed considering rhodium complexes with xantphos type ligands (Scheme 9). The correlation between the bite angles and regioselectity takes place at the transition state for the olefin insertion, which is also the regioselectity step in the catalytic cycle. Diphenylphosphino substituents play a major role in determining the selectivity. When the steric effects of the phenyl groups were removed, a notable decrease in the normal/iso ratio of the alkyl intermediates were reported. On the other hand, the orbital effects seemed to have little influence in determining the regioselectity.
 | ||
| Scheme 9 The xantphos (a) and thiaxantphos (b) ligands. | ||
Further QM/MM calculations within the ONIOM framework were carried out in order to clarify whether the olefin coordination or the hydride migratory insertion is the rate-determining step in the hydroformylation of 1-octene catalysed by Rh–xantphos systems.127 For the quantum mechanic part the B3LYP functional was employed along with double-ζ basis sets. The low level layer treated using the UFF force field.128 The axial-equatorial (ea) and the diequatorial (ee) pathways were compared and Fig. 13 summarises the calculated energy profiles.
 | ||
| Fig. 13 Potential energy profile for the early catalytic steps (until migratory insertion) of the ethylene hydroformylation catalysed by [HRh(CO)2(thiaxantphos)]. | ||
The rate constant for CO dissociation in the [HRh(CO)2(thiaxantphos)] complex is 200 h−1,129 which corresponds to a barrier of 20.1 kcal mol−1. Moreover, as based on empirical observations, the overall rate of the hydroformylation of 1-octene is estimated to be two orders of magnitude slower than the rate of the CO dissociation. Thus, the overall barrier of hydroformylation should be about 3 kcal mol−1 higher in energy than the barrier for CO dissociation. The computed barriers for the formation of the coordinatively unsaturated [HRh(CO)(thiaxantphos)] by the release of one CO is 28.7 and 21.4 kcal mol−1 for the ee and ea pathways, respectively, which is in good agreement with the experimental findings and reproduces the observations in that the dissociation of equatorial CO from the saturated ea species proceeds much faster than that from the ee complex.
As the coordination of ethylene was found to be barrierless for both pathways, the alkene coordination can be excluded as a candidate for the rate-determining step. The slower migratory insertion, which proceeds with overall barriers of 25.0 and 25.2 kcal mol−1 for the ee and ea pathways, respectively, governs the regioselectity and controls the overall activity of the catalytic cycle. However, the CO dissociation and the alkene coordination contribute to the overall barrier, therefore it is more accurate to say that the rate of Rh–xantphos catalysed hydroformylation is determined by a set of elementary steps ending in migratory insertion.
The hydroformylation of propene with rhodium complexes ligated with xantphos was studied by Landis and Uddin at the ONIOM(B3LYP/LANL2DZ:HF/LANL2MB) level of theory.130 For resting states a mixture of axial-equatorial and diequatorial isomers of [HRh(CO)2(xantphos)] was considered. The dissociation of CO from these complexes was found to be barrierless with a free energy change of about 15 kcal mol−1, somewhat lower than the experimental dissociation free energy (20 kcal mol−1).131 The computed n/iso ratio for the propene insertion into the H–Rh bond was 42:1 being in good agreement with experimental ratios of n-nonanal to i-nonanal (52:1) for 1-octene hydroformylation.131 Although the accuracy of the calculated regioselectity was satisfactory, the computations overestimated the overall activation free energies for catalytic hydroformylation. It was assumed, that much of this discrepancy between computed and experimental activation energies originated from the underestimation of propene bonding energies to the rhodium fragment.
The origin of stereoinduction by the chiral aminophosphine phosphinite (AMPP) ligands was investigated using the QM/MM approach with the BP86 functional for the inner part and with the SYBYL force field132 for the outer part of the complexes.133 It was concluded, that the alkene insertion into the Rh–H bond is the selectivity determining step, not the alkene coordination. The nonbonding weak interactions of styrene with the substituents of the phosphorous atom of the aminophosphine is responsible for the stereodifferentiation. The chirality of the AMPP backbone plays only a minor role. The two competitive equatorial/axial reaction paths show opposite asymmetric induction, therefore the rationalisation of the stereochemical outcome is not straightforward. It was assumed, that the performance of the AMPP ligands could be further increased with the combination of stereorecognition and stereohindrance by the substituents on the P atom. Furthermore, a more rigid backbone is preferential in order to promote the selective stereohindrance.
The origin of stereoinduction in the asymmetric hydroformylation of styrene catalysed by the [HRh(CO)2(BINAPHOS)] catalyst was scrutinised134 using the same level of theory as for ref. 133. The different factors governing the ligand coordination preferences and stereoinduction were evaluated. It was found that the phosphine moiety, or in general, the least basic moiety of the BINAPHOS ligand favours the equatorial coordination mode (that is, ea1 in Scheme 10). The electronic distortion of the metal fragment, however, inverses the trend to some extent, because the metal fragment is less distorted when phosphite is at the apical position. The coordination mode ea2 is less stable by 1.8 kcal mol−1, whereas the diequatorial coordination modes are higher in energy by at least 3.6 kcal mol−1. The small energy differences suggest, that small tuning of ligand properties may invert the coordination preferences. The M06 class of functionals and B97D functional were tested, but gave qualitatively the same results as did BP86, which is explained by the dominating repulsive type interactions between the ligand and the substrate.
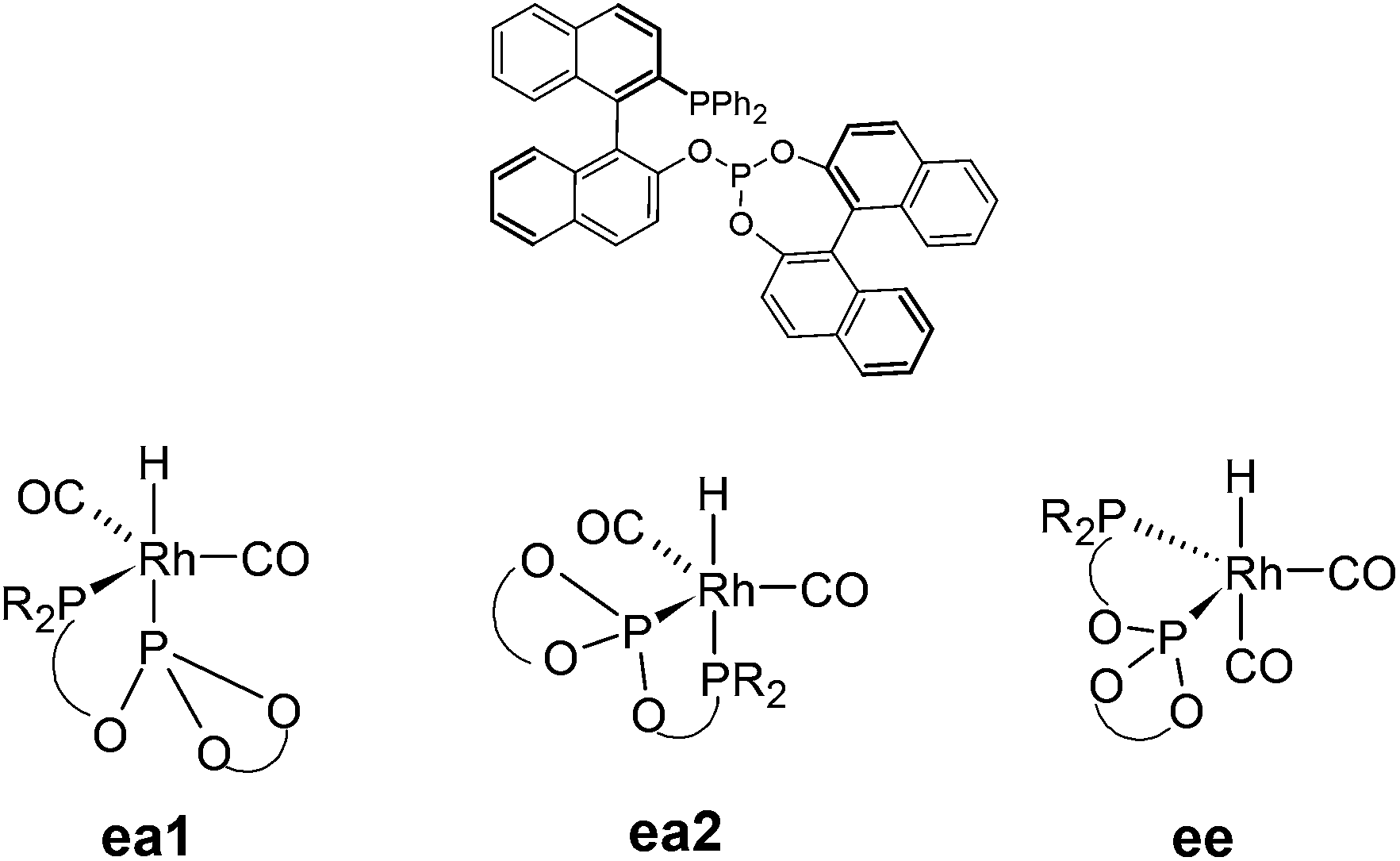 | ||
| Scheme 10 The BINAPHOS ligand (top) and its three different geometrical isomers for the hydrido rhodium carbonyl complex [HRh(CO)2(P–PO)] (bottom). | ||
On the basis of model and kinetic data, Landis and co-workers proposed a two-dimensional quadrant steric map for the interpretation of selectivity for the Rh-(S,S,S)-bisdiazaphos catalyst.135 The plane contains the Rh–H axis, and it is parallel to the alkene plane with the equatorial ligands positioned behind the plane. The axial-equatorial arrangement of the chelating ligand is assumed. With the introduction of the VW molecular descriptor, which describes the distance-weighted steric volume of the ligand and its impact on the metal centre, a new stereochemical map is proposed, which is depicted in Fig. 14. The sterically small quadrants are uncoloured, the sterically largest quadrant is dark gray, and the one between the two extrema is light gray. For path II, which is generally favoured over path I, the pro-R transition state (bottom right quadrant) is more preferred than the corresponding pro-S transition state. From Carbó and co-workers a new 3D-QSPR (quantitative structure-enantioselectivity and structure–activity relationships) model with predictive ability was developed for the asymmetric hydroformylation of styrene by Rh–diphosphines, which is based on 3D steric- and electrostatic-type interaction fields derived from DFT calculations.136
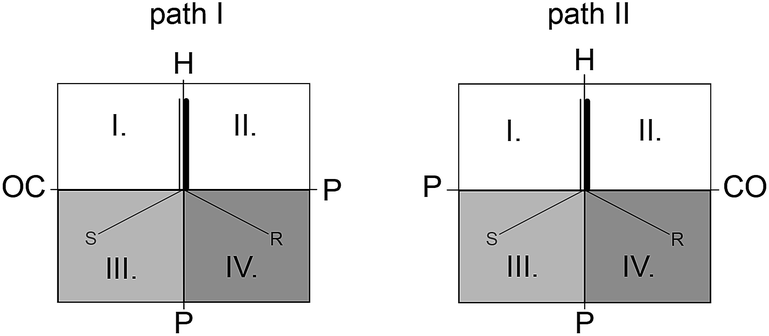 | ||
| Fig. 14 Proposed quadrant representation of the possible styrene insertion paths for BINAPHOS ligands coordinated in axial-equatorial mode. | ||
The behaviour of the π-acceptor phosphinine ligand in rhodium-catalyzed hydroformylation of alkenes was analysed using the BP86 functional and was compared to the phosphine modified catalyst precursors [HRh(CO)3(PR3)] and [HRh(CO)2(PR3)2].137 A strong preference was predicted for the 2,4,6-PC5H2Ph3 phosphinine ligand to coordinate at the equatorial site of the pentacoordinated Rh complex with the P-heterocycle perpendicular to the equatorial plane of the complex. The amount of back donation was found to be small for this π-acceptor ligand resulting in a facile rotation, and hence to a higher activity.
A new generation of wide bite angle ligands were introduced by Breit and Seiche with the in situ generation of bidentate P-donor ligands based on the self-assembly of two monodentate ligands in the coordination sphere of a metal centre through hydrogen bonding.138 The most prominent example of such supramolecular self-assembling ligands is the 6-diphenylphosphinopyridin-2-(1H)-one (6-DPPon) system which proved to give rise to an extremely active hydroformylation catalyst for terminal alkenes enabling the reactions to be carried out at ambient pressure and room temperature.
The reaction mechanism was investigated at M06/triple-ζ//B3LYP/double-ζ level of theory.139 Propene was used as a model for 1-octene. The refinement of the energies for the reaction pathway were also completed with the two-layer CCSD(T)-MP2 method on geometries obtained by DFT calculations. In this case, ethylene was considered as substrate.140
The first part of the catalytic cycle, that is the catalyst formation followed by alkene coordination and insertion, is depicted in Fig. 15. As initial complex [HRh(CO)2(6-DPPon)2] is considered, which was also detected experimentally by in situ IR spectroscopy. It was found that the transition state with the lowest free energy for the prolinear olefin insertion and the probranched olefin insertion do not start from the same alkene complex, which was also confirmed by IRC calculations. The prolinear alkene complex is higher in energy, and therefore the resulting free energy of activation value is smaller. The n/iso ratio, calculated from the two different activation free energy values, is in good agreement with the experimental observations. The computed free energy surface shows that the prolinear alkyl formation step is exothermic by −6.2 kcal mol−1, whereas the probranched olefin insertion is slightly endothermic by 1.4 kcal mol−1.
 | ||
| Fig. 15 Free energy surface for the first part of the catalytic cycle (that is the propene coordination and insertion followed by CO coordination) at the M06 level of theory. The linear pathway is black, whereas the branched pathway is designated with gray colour. | ||
Tonks, Froese and Landis employed the (S,S,S)-bis-diazaphos (BDP) ligand for the rhodium-catalysed hydroformylation of styrene.141 The Rh-BDP system allows the reaction to be performed under extremely low pressures of syngas, even 10−2 bar as partial pressure. At partial pressures lower than 0.7 bar, the catalytic system is selective for the linear regioisomer, 3-phenyl propanal, whereas at high pressure, it is highly regio- and enantioselective for the branched (R)-2-phenylpropanal. Under low-pressure conditions the qualitative kinetic data suggest that the catalytically active species accumulate as either 4-coordinate Rh(bisphosphine)(CO)(alkyl) or 5-coordinate Rh(bisphosphine)(CO)(alkyl)(styrene) complexes (Scheme 11.) Thus, simple changes in the concentration of CO change the rate law and hence the regio- and enantioselectivity. The competitive binding between CO and styrene was investigated with M06 functional in combination with triple-ζ basis sets. Comparing the free energies, neither binding is particularly strong: −3.8 and −2.4 kcal mol−1 for CO, and for styrene, respectively. At very low styrene and CO concentration it is likely, that the coordinatively unsaturated Rh(bisphosphine)(CO)(alkyl) complex is favoured. The inhibitory effect of styrene and first-order dependence of CO on the reaction rate at low pressure suggests that the rate-limiting process involves styrene dissociation followed by coordination and insertion of carbon monoxide affording the Rh-acyl intermediate.
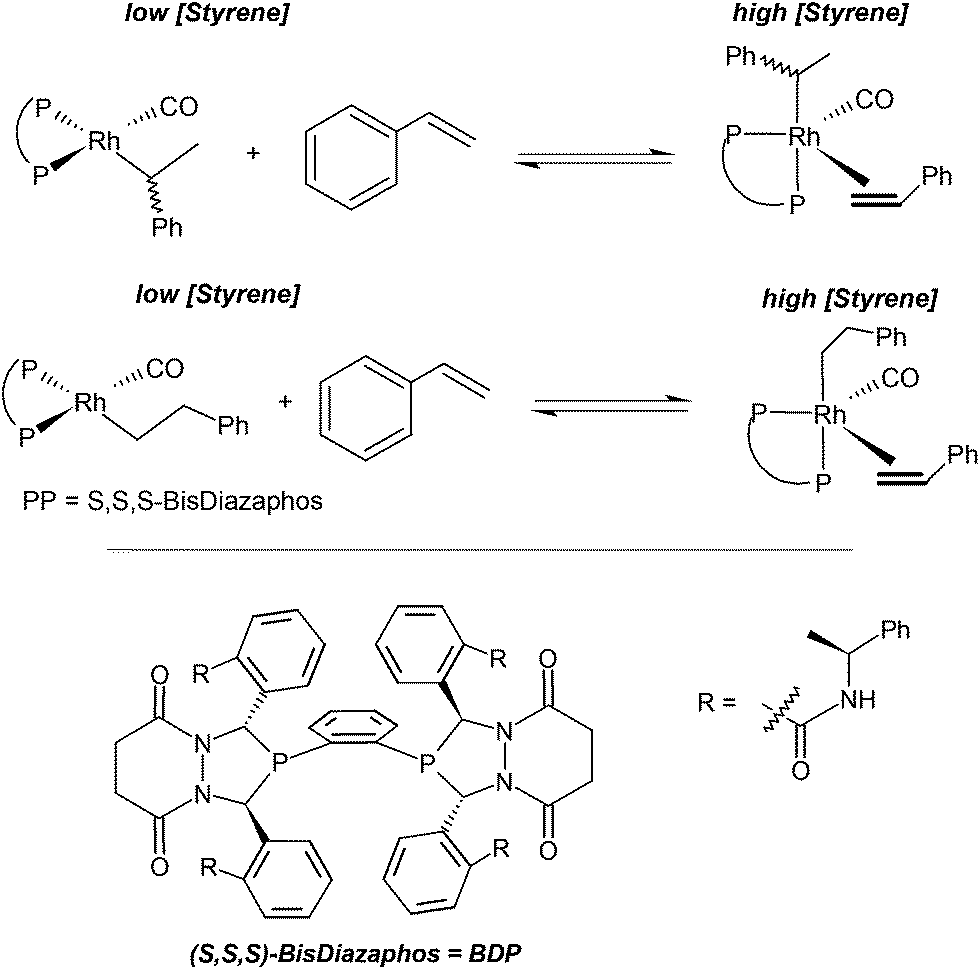 | ||
| Scheme 11 Kinetic scheme for Rh(BDP)-catalyzed styrene hydroformylation under low pressure conditions. | ||
The catalytic cycle of the rhodium-catalysed hydroformylation of 1,3-butadiene with a triptycene-derived bisphophite ligand was elucidated with DFT calculations.142 Only monohydroformylation, leading to unsaturated aldehydes, was scrutinised. Two dominant pathways were highlighted leading to 3-pentenal and 4-pentenal, which experimentally also are the main primary products. The terminal aldehyde (4-pentenal) is preferential, because with catalyst studied by the authors it can be exclusively converted to the bis-hydroformylation product adipic aldehyde.
As seen in Fig. 16 the iso-pathway, which is lower by 1.4 kcal mol−1 than that for the n-pathway in terms of free energy, enables the formation of an η3-crotyl complex, which opens up an undesirable exit channel within the catalytic cycle, resulting in 3-pentenal, which does not lead to adipic aldehyde. Apart from the kinetic reason, the formation of 3-pentenal is also preferred thermodynamically because of the more stable internal double bond compared to the terminal double bond.
 | ||
| Fig. 16 Free energy profiles of the normal and iso pathways for the first part of butadiene hydroformylation (that is the olefin coordination and insertion followed by CO coordination) at the M06 level. The normal pathway is gray, whereas the iso pathway is designated with black colour. | ||
The prediction of selectivity is not a straightforward task for this type of reaction. Because of the stable η3-crotyl intermediate the migratory insertion step is predicted to be irreversible. However, for the n-pathway the olefin insertion may be partly reversible because the aldehyde reductive elimination transition state is almost equally high in free energy, thus β-elimination of the Rh-alkyl intermediate may take place depending on the reaction conditions.
The mechanism for the hydroformylation of phosphinobutene catalysed by the heterobinuclear complex (CO)4Cr(μ-PH2)2RhH(CO)(PH3) was studied by Tang et al.143 in order to model the hydroformylation of phosphinobutene CH2CHCH2CH2PPh2 catalysed by the Rh(I) complex (CO)4Cr(μ-PPh2)2RhH(CO)(PPh3).144 For the catalytically active species two isomers were proposed from which the one with hydrido and CO ligands on Rh are in trans position is somewhat less stable, but more active in terms of olefin coordination and insertion. The Rh–Cr distance (2.848 Å) and the Wiberg bond index (0.2) suggest a bonding interaction between the two transition metal atoms. Furthermore, the HOMO-3 orbital (see Fig. 17) indicates an interaction between the 4d orbitals of rhodium and the 3d orbitals of chromium. Molecular orbital analysis revealed that the LUMO of this complex contains 11.5% dx2−y2 orbital of the Rh atom, which is a good acceptor orbital for the π electrons of olefin.
 | ||
| Fig. 17 HOMO-3 (left) and LUMO (right) orbitals of the heterobinuclear complex (CO)4Cr(μ-PH2)2RhH(CO)(PH3). | ||
It was concluded, that the bimetallic Rh–Cr-catalyzed hydroformylation follows a chelate associative mechanism. The branched product is favoured with a regioselectivity of almost 100%. The Cr serves as an orbital reservoir in the olefin addition and insertion steps via the variation of the orbital interaction between chromium and rhodium atoms.
5. Other metals as catalysts
5.1. Hydroformylation by platinum containing systems
The platinum/tin-catalysed hydroformylation of olefins was first reported in the literature by Hsu and Orchin in 1975,145 and Schwager and Knifton in 1976.146 Soon, this catalytic system was successfully applied for asymmetric hydroformylation employing various chiral phosphines. Remarkable enantioselectivities were observed both for “preformed” PtCl(SnCl3)(diphosphine) catalysts and for in situ PtCl2(diphosphine) + tin(II) chloride systems.147–155 Apart from SnCl2 tin(II) fluoride has also successfully been employed as cocatalyst.156 Platinum/tin systems were also reported as suitable catalysts for the regioselective hydroformylation of functionalised internal alkenes.7It is generally accepted that the initial step of the reaction is the coordination of olefin onto the hydrido complex157 formed from the analogous chloro complex under hydrogen atmosphere. This species contains the SnCl3 ligand directly coordinated to platinum. As the chloro complex is usually the precursor complex for the catalytic reaction, no surprise that the first computational studies aimed to explore the structure and isomerisation of the [PtCl(SnCl3)(PH3)2] model system.
The insertion reaction of SnCl2 into the Pt–Cl bond was studied by Rocha and de Almeida.158 The geometries were optimised at the HF level, whereas the energy refinements were made at MP2 level of theory. The trans-[PtCl(SnCl3)(PH3)2] complex was found to be more stable by 7.0 kcal mol−1 than the corresponding cis isomer. Interestingly, the energy of the transition state describing the SnCl2 insertion (see Fig. 18, left) was obtained to be lower by 4.4 kcal mol−1 than the reactants together. When solvent effects were taken into considerations, the cis isomer appeared to be more stable by 2.1 kcal mol−1 at MP2 level employing the IPCM159 solvation model in accord with the experimental studies of Chatt and Wilkins.160 The isomerisation was found to undergo via the transition state depicted in Fig. 18, right with a barrier over 30 kcal mol−1.161
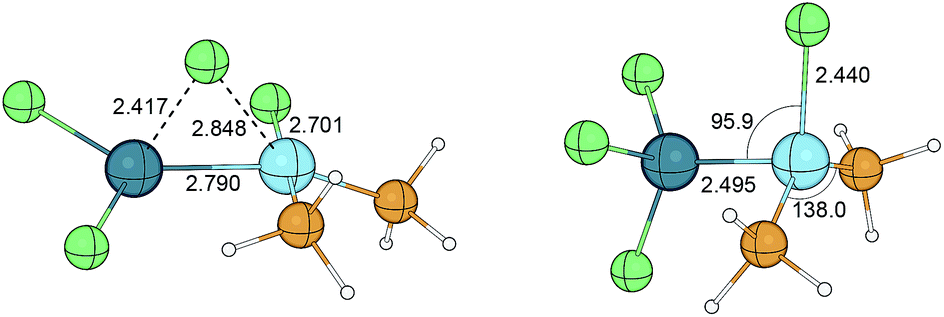 | ||
| Fig. 18 Transition states describing the SnCl2 insertion into the Pt–Cl bond in cis-[PtCl(SnCl3)(PH3)2] (left) and the isomerisation between cis- and trans-[PtCl(SnCl3)(PH3)2] (right). Bond distances are given in Å, angles in degree. | ||
The formation of cis-[PtCl(SnCl3)(PH3)2] was investigated at the B3LYP/LANL2DZp level in order to resolve the role of imidazolium-based ionic liquids containing chlorostannate anions.162 The energy of transition state representing the SnCl2 is by 2.8 kcal mol−1 lower in energy, than the reactants cis-[PtCl2(PH3)2] and SnCl2 together, in accord with the HF/MP2 calculations.158 The “mystery of negative barrier” can be resolved, however, with the introduction of the strongly bound adduct of SnCl2 with the dichloro platinum complex which is exothermic by −18.7 kcal mol−1 (see Fig. 19). The coordination of SnCl2 toward one of the chloro ligands results in a slight elongation of its bond distance with platinum. The insertion takes place via a three-centre transition state with a barrier of 15.9 kcal mol−1 affording complex cis-[PtCl(SnCl3)(PH3)2]. The process was found to be exothermic by −13.1 kcal mol−1.
 | ||
| Fig. 19 Reaction pathway for the addition of SnCl2 to cis-[PtCl2(PH3)2]. Bond distances are given in Å. | ||
Based on HP NMR studies it was assumed that for some phosphines the platinum-catalysed hydroformylation can take place via an ionic mechanism, when the trichlorostannate ligand, which is a good leaving group, acts as an anion, thus can readily be replaced by other ligands, such as CO.163 The insertion of ethylene in cationic platinum–hydrido–diphosphine complexes [HPt(PX3)2]+ (where X = H, Cl, Me) was investigated at HF, MP2, BLYP, and BPW91 levels of theory.164 The barrier for the olefin insertion was found to be small, indicating that this is not the rate-limiting step for cationic catalysts. Car–Parrinello molecular dynamics calculations94 were also performed. For X = H the insertion does not take place instantaneously whereas the time scales for completion of the insertion in the presence of PCl3 and PMe3 is roughly 100 fs in accord with the static calculations which predict smaller barriers for those cases.
The insertion of propene into the Pt–H bond in the cationic complex [HPt(PX3)2]+ (where X = H, F, Me) was studied at HF, MP2, and B3PW91 levels of theory.165 Small insertion barrier was found for the cases X = H, Me (less than 2.5 kcal mol−1), whilst the process was found almost barrierless for the PF3 containing system. HF calculations predicted notably higher activation energies which was attributed to the lack of correlation effects.
The hydroformylation of styrene in the presence of dialkyl/diaryl–Pt(BDPP) complexes (where BDPP stands for (2S,4S)-2,4-bis(diphenylphosphino)pentane) combined with strong Lewis-acids such as B(C6F5)3 or BF3 is a rare example for tin(II)-halide-free Pt-catalysed hydroformylation. BP86/PCM calculations revealed that the formation of the presumed cationic precursor takes place via an SN2-like reaction.166
The hydroformylation of styrene in the presence of PtCl(SnCl3)[(2S,4S)-BDPP] precursor shows a strong temperature dependence with the preference of formation of (S)-2-phenyl propanal at low temperature and the (R) enantiomer at high temperature.112,156,168 The reversal temperature of enantioselectivity depends on the Hammett constant (σp) of the functional group in 4-substituted styrenes, therefore the effect of the para-substituent on styrene was elucidated using the simple model compounds HPt(PH3)2(SnCl3)(olefin) and the PBEPBE functional (Fig. 20).167 The electrostatic potential at nuclei (EPN) was chosen as quantum chemical descriptor which is defined according to eqn (3).
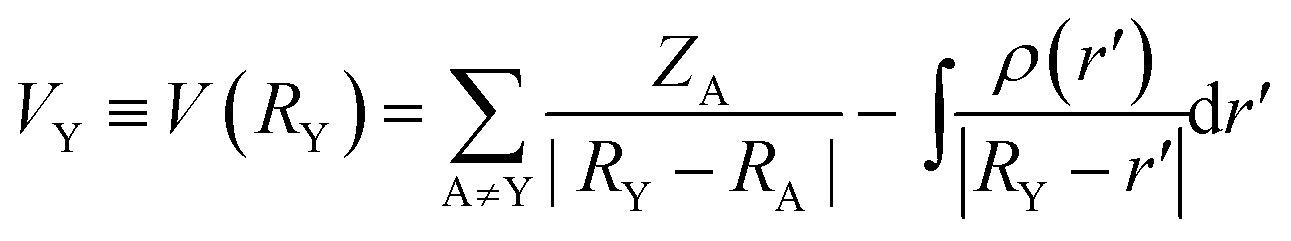 | (3) |
 | ||
| Fig. 20 Computed structure of styrene and Pt–hydride complexes bounding para-substituted styrenes. Bond distances are given in Å. Reproduced with permission from ref. 167. Copyright 2014 American Chemical Society. | ||
 | ||
| Fig. 21 Relationship between the EPN or Pt in complexes [HPt(PH3)2(SnCl3)(olefin)] and the ZPE-corrected binding energies of para-substituted styrenes. Reproduced with permission from ref. 167. Copyright 2014 American Chemical Society. | ||
The mechanism of Pt/Sn catalysed hydroformylation was scrutinised by Rocha and de Almeida by investigating the elementary steps employing simple model compounds. The insertion of ethylene into the Pt–H bond was investigated at the MP4(SDQ)//MP2 level of theory.170 The activation energy was found to be only 11.8 kcal mol−1, and the β-hydrogen elimination two times greater, indicating that the insertion step was irreversible. It was also concluded that the SnCl3 ligand weakens the Pt–H bond during the insertion and stabilises the pentacoordinated intermediates. In the absence of the trichlorostannate ligand the migratory insertion proceeds with an energy barrier of 33.9 kcal mol−1, which explains why the [PtCl2(P)2] type of complexes show no catalytic activity during the hydroformylation of olefins without the addition of tin(II)-halides. The transition structure accounting for the olefin insertion is depicted in Fig. 22 (top left).
 | ||
| Fig. 22 Computed structures for transition states for modeling the olefin insertion (top left), CO insertion (top right) and aldehyde reductive elimination (bottom) calculated at the MP4(SDQ)//MP2 level. Bond distances are given in Å. | ||
The MP4(SDQ)//MP2 level was employed for the investigation of the migratory insertion of the carbonyl ligand into the platinum–methyl carbon bond.171 The reaction proceeds via a three centre transition state with a trigonal-bipyramidal like structure. Intrinsic reaction coordinate (IRC) calculations172 showed that this reaction occurs with a progressive migratory insertion of the carbonyl ligand into the Pt–Cmethyl bond, followed by an intramolecular rearrangement to generate the square-planar Pt–acyl product. The carbonyl insertion step is endothermic by 1.9 kcal mol−1 and takes place with an activation energy of 26.4 kcal mol−1. It should be noted, however, that pure MP2 calculations predicted notably higher endothermicity (8.0 kcal mol−1). NBO calculations173 showed, that the participation of d-orbitals on the Sn atom is negligible when forming the Pt–Sn bond. The transition structure accounting for the CO insertion is depicted in Fig. 22 (top right).
The hydrogenolysis process, namely the H2 oxidative addition and aldehyde reductive elimination was investigated at the MP4(SDQ)//MP2 level as well.174 The total energy barrier is 33.9 kcal mol−1 which makes clear why the hydrogenolysis is the rate-determining step for some Pt/Sn/phosphine systems. Closer examination of this process shows, that the dihydrogen activation is faster, than the aldehyde elimination, and a relatively stable dihydrido Pt(IV) complex is formed during the reaction. The Charge Decomposition Analysis (CDA)175 revealed that the addition proceeds with an increasing electron donation from the metallic fragment to the antibonding σ* orbital of the incoming H2 molecule.
The elimination of the aldehyde takes place involving either the cis or trans hydrido ligand with respect to SnCl3 in the Pt(IV) complex. The two transition states accounting for this elementary step are depicted in Fig. 22 (bottom). The axial-equatorial transition state, that is, when SnCl3 is trans to the reacting hydrido ligand, is lower in energy by 3.2 kcal mol−1 in comparison to the diequatorial transition structure. This result also emphasizes the role of the trichlorostannate ligand, which has an advantageous influence on the rate of hydrogenolysis as well.
The entire catalytic cycle for the hydroformylation of propene was also investigated by Rocha et al. employing DFT methods.176 Several GGA, hybrid GGA and meta-GGA functionals were tested for the energetics of olefin insertion and compared to CCSD(T) values. It was found that the BP86 and GPW91 functionals gave the best estimation for the coordination of ethylene to the metallic fragment, whereas all DFT methods somewhat underestimated the barrier for insertion. Among the elementary steps the hydrogenolysis was found as rate-determining with an activation energy of 21 kcal mol−1. The BP86/cc-pVTZ/SBKJC results showed that the pathway leading to the linear aldehyde was preferred.
The mechanism of propene hydroformylation in the presence of model catalysts cis-[PtH(SnCl3)(PH3)2] and trans-[PtH(SnCl3)(PH3)2] was investigated by Bedekovits and co-workers at the B3LYP level of theory.177 The regioselectivity was found to be determined in the olefin insertion step. When solvation corrections were employed the computed ratio of the linear regioisomer, n-butanal was predicted to be 83% being in very good agreement with the regioselectity reported by Schwager and Knifton (85%).146
In order to get deeper insight into the electronic structure of species of olefin insertion along the reaction path IRC calculation was completed and the partial charges of atoms of higher importance were determined during the reaction (Fig. 23). The NPA charge178 of the Pt centre showed a gradual decrease as it takes away some electron density from the hydride ligand. The charge of the H atom does not change until the transition state is reached but it shows a sudden increase while migrating to the carbon atom. The electron density of the former hydride ligand is mainly transferred to the methine carbon (C1) while the C–H bond is formed. Approaching the transition state the electron density of one of the phosphorous atoms (P1) decreases, while that of P2 increases, thus the two ligands provide a balancing effect for the charge distribution in the coordination sphere of platinum.
 | ||
| Fig. 23 Intrinsic reaction coordinate (IRC) of the propene insertion into the Pt–H bond of the olefin adduct resulting in the cis linear propyl complex (top) and the variation of the natural charges of selected atoms along the reaction coordinate (bottom). Reproduced with permission from ref. 177. Copyright 2010 Elsevier. | ||
The Quantum Theory of Atoms In Molecules (QTAIM) analysis179 revealed ring critical points (RCPs) in all Pt–olefin adducts. The charge distribution of the electron density in the Pt–C–C plane is strongly influenced by the SnCl3 ligand. In equatorial position it shifts the RCP significantly closer to the Pt–C bond adjacent to the phosphine ligand (Fig. 24).
 | ||
| Fig. 24 Laplacian distribution ∇2ρ of the [PtH(SnCl3)(PH3)2(propene)] adducts with the SnCl3 ligand in equatorial (left) or in axial (right) positions. Green dots indicate bond critical points, whereas red dots indicate ring critical points. | ||
The effect of the SnCl3 ligand in the dihydrido–acyl complexes was investigated as well. The trans arrangement of the acyl group and SnCl3 results in a more even electron density distribution which is reflected in the large energy difference (13.9 kcal mol−1) in favour of the trans complex (Fig. 25, left).
 | ||
| Fig. 25 Computed structures of Pt–dihydrido–acyl complexes with selected bond lengths (given in Å). Natural charges are written in italics. Reproduced with permission from ref. 177. Copyright 2010 Elsevier. | ||
The mechanism of propene hydroformylation promoted by complex [PtH(SnCl3)(PH3)2] was investigated by Rocha et al. employing the BP86 functional and solvation corrections.180 In agreement with previous studies176,177 the regioselectity was found to be determined at the olefin insertion step. Employing the kinetic model based on concurrent transition states depicted in Fig. 26 a linear regioselectity of 95% was computed, which is somewhat higher than reported by Schwager and Knifton.146 The aldehyde reductive elimination was predicted as the slowest step with an activation free energy of 18.1 kcal mol−1.
 | ||
| Fig. 26 Olefin insertion transition states leading to linear (left) and branched (right) alkyl complexes. Bond distances are given in Å. | ||
The Pt/Sn catalysed asymmetric hydroformylation of styrene in the presence of the model complex PtH(SnCl3)(chiraphosH) (chiraphosH = (2S,3S)-(2,3)-diphosphinobutane) has been investigated by means of DFT calculations using the PBEPBE functional with solvation corrections employing the CPCM182 model.181 The inspection of the potential energy hypersurfaces for the three preferred reaction channel show, that olefin coordination followed by the olefin insertion step is the rate-determining in all cases with activation barriers of 21.0, 20.5, and 19.7 kcal mol−1 for the linear, S and R pathways, respectively. In the presence of CO there is a strong energetic preference toward the formation of the acyl complexes, which undergo a relatively fast H2 oxidative addition step followed by the somewhat slower aldehyde reductive elimination (Fig. 27).
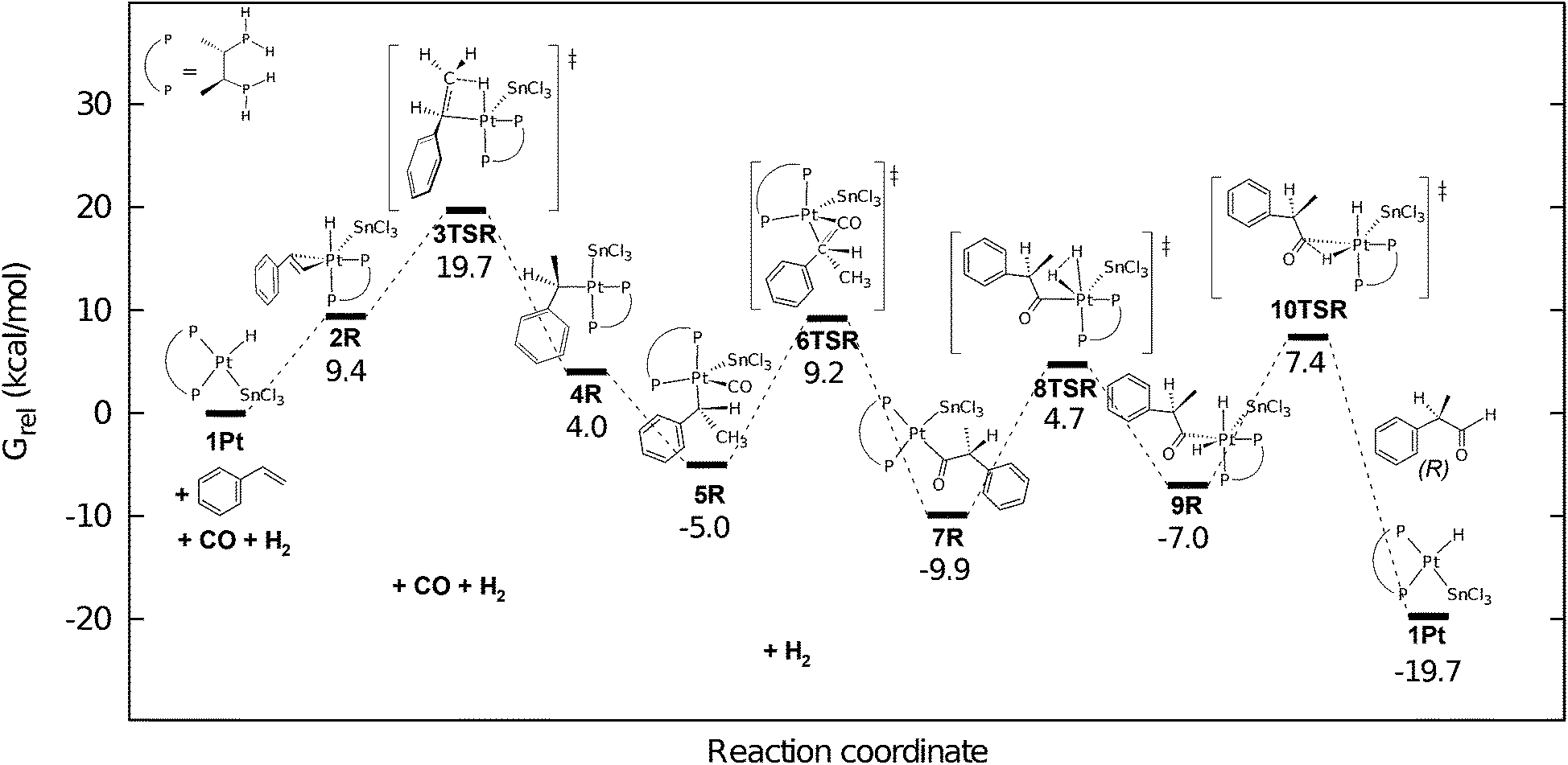 | ||
| Fig. 27 Free energy profile for the formation of (R)-2-phenyl-propanal catalysed by [PtH(SnCl3)(chiraphosH)] Reproduced with permission from ref. 181. Copyright 2013 American Chemical Society. | ||
The oxidative addition of H2 may take place via a transition state where the coordinating hydrogens are perpendicular to the Pt−Cacyl bond (perpendicular arrangement), or via a transition structure where the hydrogen atoms and the acyl carbon lie in the same plane (parallel arrangement) (Scheme 12). As a consequence, octahedral complexes are formed keeping the same orientation of hydride ligands to those in the corresponding transition states. The difference in free energy between the two transition states is almost preserved indicating a strong energetic preference in favour of the perpendicular arrangement.
 | ||
| Scheme 12 Possible pathways for hydrogen addition and aldehyde reductive elimination resulting in the formation of the branched (R) 2-phenyl-propanal. Free energy values are given in kcal mol−1. | ||
The elimination of aldehyde from the more stable dihydrido complex can undergo following two pathways involving the hydrido ligand either in trans or cis position with respect to SnCl3. The barrier, associated with the first pathway is exactly the half (13.1 kcal mol−1) in comparison to the second one taking place through a transition state in which the axial hydrido ligand coordinates to the acyl carbon (26.2 kcal mol−1).
The distribution of aldehyde products was found to be unequivocally determined during the migratory insertion of styrene. Therefore, the regio-, as well as the enantioselectivity can be determined via the relative rate of the concurrent olefin insertion pathways. The coordination of styrene towards the initial hydrido complex may result, in principle, 8 olefin adducts, with trigonal bipyramidal structure. The structural difference of the adducts stems from the two possible locations of the trichlorostannate ligand (axial, or equatorial) and from the orientation of the phenyl group of styrene which can be placed in either of the four quadrants, determined by the position of the hydrogen, the tin and the two phosphorous atoms. The subsequent migratory insertion may proceed by the rotation of the olefin ligand in two directions increasing the number of attainable transition states to 16. Some combinations lead to the very same ones, thus, the real number of transition states describing the migratory insertion of styrene is 11, which are depicted schematically in Fig. 28.
 | ||
| Fig. 28 Simplified representation of transition states governing the chiral discrimination as well as regioselectivity. Activation free energy values are given in kcal mol−1. Percentages obtained at PBEPBE level are written in black whereas MP4(SDQ) results are in grey. Reproduced with permission from ref. 181. Copyright 2013 American Chemical Society. | ||
The relative amount of two aldehyde isomers, for instance that of (R)-2-phenyl-propanal compared to that of (S)-2-phenyl-propanal can be expressed according to eqn (4) taking into account all transition states accounting for the formation of branched alkyl complexes.
 | (4) |
Thus, for enantioselectivity ee = 51%, with the preference of the R enantiomer (47% at the MP4(SDQ)//PBEPBE level), has been predicted which is in excellent agreement with the enantioselectivity (45%) obtained experimentally by Consiglio and co-workers.183 Somewhat less satisfactory result has been obtained, however, for the regioselectivity which was calculated to be 86%, in favor of the branched isomers, as opposed to the experimental value of 62%.
The catalytic cycle of ethylene hydroformylation by organoplatinum hydride complexes with hydrophosphoryl ligands was investigated by Ustynyuk and co-workers employing the PBE functional.184 The strong hydrogen bond between the two ligand in cis position enables the formation of a quasi-bidentate ligand whose rigidity may enhance the linear regioselectity.
5.2. Hydroformylation by ruthenium containing systems
Ruthenium-catalysed hydroformylation experiments often carried out in the presence of the homoleptic complex Ru3(CO)12 or with anionic complexes [HRu(CO)4]−, [HRu3(CO)11]− or [H3Ru4(CO)12]−.185 Active catalytic systems were also reported with triruthenium dodecacarbonyl in combination with chelating N-donor ligands, such as 2,2′-bipyridine, or 2,2′-bipyrimidine.186,187The catalytic activity of ruthenium carbonyl complexes with 2-substituted pyrazine ligands in the hydroformylation of 1-hexene was studied and the elementary steps of the reaction was scrutinised using the B3PW91 hybrid functional.188 During the reaction both aldehydes and alcohols are formed revealing that hydroformylation is followed by the hydrogenation of the aldehydes.
The proposed mechanism is depicted in Scheme 13. In the first step the alkene coordinates to the pentacoordinated ruthenium complex (step 1) followed by decarbonylation (step 2). In step 3 dihydrogen fills the vacancy affording the dihydrogen intermediate. Similar Ru–dihydrogen complexes are known from the literature.189 The forth step involves the intramolecular transfer of hydrogen to the alkene to form the HRu(alkyl) intermediate. In the next step (step 5) carbonyl insertion takes place followed by the generation of the aldehyde moiety (step 6). An alternative pathway involves the formation of a metal dihydride intermediate instead of the intramolecular hydrogen transfer (step 7). Due to the high relative energy of the dihydrido complex this route is not favoured even though the following hydrogenation of alkene (step 8) is an exothermic step.
 | ||
| Scheme 13 Possible routes for the hydroformylation of ethylene catalysed by [Ru(CO)3Cl2(pz)]. | ||
For the analogous catalytic system containing pyridine instead of pyrazine the presence of complex [Ru(CO)3Cl2] was predicted as catalytically active species. The loss of pyridine ligand in the initiation step was corroborated experimentally by using NMR and IR spectroscopy. In contrast to the pyrazine containing system the preferred pathway is the formation of the dihydrogen complex followed by olefin coordination and insertion. The subsequent CO insertion and aldehyde elimination steps are both exothermic.190
6. Conclusions
The hydroformylation reaction catalysed by various transition metal complexes follows a common general scheme including olefin coordination, its migratory insertion into the metal–hydrogen bond, CO coordination and subsequent insertion, H2 oxidative addition and aldehyde reductive elimination. Computational studies may help to uncover the rich mechanistic picture, with parallel multistep pathways often involving similar barriers. The main challenge is the scrutiny of the reaction features for specific sets of catalysts and substrates. With the utilization of more and more advanced techniques computational chemistry will be able to advance towards a very powerful tool for the design of more efficient and highly selective catalytic systems.Acknowledgements
The author wishes to thank the financial support of the János Bolyai Research Grant from the Hungarian Academy of Sciences. The support of the Supercomputer Center of the Hungarian National Information Infrastructure Development (NIIF) Program is also acknowledged.References
- O. Roelen, Chemische Verwertungsgesellschaft mbH, Oberhausen, German Pat. DE 849548, 1938/1952; O. Roelen, Chemische Verwertungsgesellschaft mbH, Oberhausen, US Pat., 2327066, 1943.
- F. Hebrard and P. Kalck, Chem. Rev., 2009, 109, 4272–4282 CrossRef CAS PubMed.
- Rhodium Catalyzed Hydroformylation, ed. P. W. N. M. van Leeuwen and C. Claver, Kluwer Academic Publishers, Dordrecht, Netherlands, 2000 Search PubMed.
- G. T. Whiteker and C. J. Cobley, Top. Organomet. Chem., 2012, 42, 35–46 CrossRef CAS.
- A. M. Trzeciak and J. Ziółkowski, Coord. Chem. Rev., 1999, 190–192, 883–900 CrossRef CAS.
- M. Diéguez, O. Pàmies and C. Claver, Tetrahedron: Asymmetry, 2004, 15, 2113–2122 CrossRef PubMed.
- R. van Duren, J. I. van der Vlugt, H. Kooijman, A. L. Spek and D. Vogt, Dalton Trans., 2007, 1053–1059 RSC.
- L. Wu, I. Fleischer, R. Jackstell, I. Profir, R. Franke and M. Beller, J. Am. Chem. Soc., 2013, 135, 14306–14312 CrossRef CAS PubMed.
- K. Tominaga and Y. Sasaki, J. Mol. Catal. A: Chem., 2004, 220, 159–165 CrossRef CAS PubMed.
- I. Piras, R. Jennerjahn, R. Jackstell, A. Spannenberg, R. Franke and M. Beller, Angew. Chem., Int. Ed., 2011, 50, 280–284 CrossRef CAS PubMed.
- J. Palágyi and L. Markó, J. Organomet. Chem., 1982, 236, 343–347 CrossRef.
- H. Marrakchi, J.-B. N. Effa, M. Haimeur, J. Lieto and J.-P. Aune, J. Mol. Catal., 1985, 30, 101–109 CrossRef CAS.
- A. Fukuoka, T. Kimura, L.-F. Rao and M. Ichikawa, Catal. Today, 1989, 6, 55–62 CrossRef CAS.
- R. Franke, D. Selent and A. Boerner, Chem. Rev., 2012, 112, 5675–5732 CrossRef CAS PubMed.
- P. Eilbracht, L. F. ärfacker, C. Buss, C. Hollmann, B. E. Kitsos-Rzychon, C. L. Kranemann, T. Rische, R. Roggenbuck and A. Schmidt, Chem. Rev., 1999, 99, 3329–3366 CrossRef CAS PubMed.
- F. Agbossou, J.-F. Carpentier and A. Mortreux, Chem. Rev., 1995, 95, 2485–2506 CrossRef CAS.
- C. Botteghi, S. Paganelli, A. Schionato and M. Marchetti, Chirality, 1991, 3, 355–369 CrossRef CAS PubMed.
- S. Gladiali, J. C. Bayón and C. Claver, Tetrahedron: Asymmetry, 1995, 6, 1453–1474 CrossRef CAS.
- A. Gual, C. Godard, S. Castillón and C. Claver, Tetrahedron: Asymmetry, 2010, 21, 1135–1146 CrossRef CAS PubMed.
- J. Pospech, I. Fleischer, R. Franke, S. Buchholz and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 2852–2872 CrossRef CAS PubMed.
- (a) F. Ungváry, Coord. Chem. Rev., 1995, 141, 371–493 CrossRef; (b) F. Ungváry, Coord. Chem. Rev., 1996, 147, 547–570 CrossRef; (c) F. Ungváry, Coord. Chem. Rev., 1997, 160, 129–159 CrossRef; (d) F. Ungváry, Coord. Chem. Rev., 1997, 167, 233–260 CrossRef; (e) F. Ungváry, Coord. Chem. Rev., 1998, 170, 245–281 CrossRef; (f) F. Ungváry, Coord. Chem. Rev., 1999, 188, 263–296 CrossRef; (g) F. Ungváry, Coord. Chem. Rev., 2001, 213, 1–50 CrossRef; (h) F. Ungváry, Coord. Chem. Rev., 2001, 218, 1–41 CrossRef; (i) F. Ungváry, Coord. Chem. Rev., 2002, 228, 61–82 CrossRef; (j) F. Ungváry, Coord. Chem. Rev., 2003, 241, 295–312 CrossRef; (k) F. Ungváry, Coord. Chem. Rev., 2004, 248, 867–880 CrossRef PubMed; (l) F. Ungváry, Coord. Chem. Rev., 2005, 249, 2946–2961 CrossRef PubMed; (m) F. Ungváry, Coord. Chem. Rev., 2007, 251, 2072–2086 CrossRef PubMed; (n) F. Ungváry, Coord. Chem. Rev., 2007, 251, 2087–2102 CrossRef PubMed.
- T. Kégl, Modern Carbonylation Methods, ed. L. Kollàr, Wiley-VCH, Weinheim, Germany, 2008, pp. 161–198 Search PubMed.
- S. Niu and M. B. Hall, Chem. Rev., 2000, 100, 353–406 CrossRef CAS PubMed.
- M. Torrent, M. Solà and G. Frenking, Chem. Rev., 2000, 100, 439–494 CrossRef CAS PubMed.
- A. Dedieu, Chem. Rev., 2000, 100, 543–600 CrossRef CAS PubMed.
- C. E. Webster and M. B. Hall, Coord. Chem. Rev., 2003, 238–239, 315–331 CrossRef CAS.
- A. C. Tsipis, Coord. Chem. Rev., 2014, 272, 1–29 CrossRef CAS PubMed.
- K. Raghavachari and J. A. Pople, Int. J. Quantum Chem., 1978, 14, 91–100 CrossRef.
- G. D. Purvis III and R. J. Bartlett, J. Chem. Phys., 1982, 76, 1910–1918 CrossRef PubMed.
- P. E. M. Siegbahn, Advances in Chemical Physics, ed. S. A. Rice and I. Prigogine, John Wiley & Sons, New York, 1996, vol. XCIII, p. 333 Search PubMed.
- C. J. Cramer and B. A. Smith, J. Phys. Chem., 1996, 100, 9664–9670 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- W. R. Rocha, E. S. Xavier, J. C. S. Da Silva, R. P. Dias, H. F. Dos Santos and W. B. de Almeida, J. Theor. Comput. Chem., 2012, 11, 297–312 CrossRef CAS.
- C. Adamo and V. Barone, J. Chem. Phys., 1998, 108, 664–675 CrossRef CAS PubMed.
- X. Feng, J. Gu, Y. Xie, R. B. King and H. F. Schaefer, J. Chem. Theory Comput., 2007, 3, 1580–1587 CrossRef CAS.
- B. I. Dunlap, J. Chem. Phys., 1983, 78, 3140–3142 CrossRef CAS PubMed.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS.
- J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822–8824 CrossRef.
- J. P. Kenny, R. B. King and H. F. Schaefer, Inorg. Chem., 2001, 40, 900–911 CrossRef CAS.
- J. M. Tukacs, D. Király, A. Strádi, G. Novodarszki, Z. Eke, G. Dibó, T. Kégl and L. T. Mika, Green Chem., 2012, 14, 2057–2065 RSC.
- Y. Zhao and D. Truhlar, J. Chem. Phys., 2006, 125, 194101–194118 CrossRef PubMed.
- Y. Zhao and D. G. Truhlar, Acc. Chem. Res., 2008, 41, 157–167 CrossRef CAS PubMed.
- A. D. Kulkarni and D. G. Truhlar, J. Chem. Theory Comput., 2011, 7, 2325–2332 CrossRef CAS.
- L. Goerigk and S. Grimme, Phys. Chem. Chem. Phys., 2011, 13, 6670–6688 RSC.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- X. Xu and D. G. Truhlar, J. Chem. Theory Comput., 2012, 8, 80–90 CrossRef CAS.
- B. S. Narendrapurapu, N. A. Richardson, A. V. Copan, M. L. Estep, Z. Yang and H. F. Schaefer, J. Chem. Theory Comput., 2013, 9, 2930–2938 CrossRef CAS.
- I. Wender, H. W. Sternberg and M. Orchin, J. Am. Chem. Soc., 1953, 75, 3041–3042 CrossRef CAS.
- D. Antolovic and E. R. Davidson, J. Am. Chem. Soc., 1987, 109, 977–985 CrossRef CAS.
- D. Antolovic and E. R. Davidson, J. Am. Chem. Soc., 1987, 109, 5828–5840 CrossRef CAS.
- E. A. McNeill and F. R. Scholer, J. Am. Chem. Soc., 1977, 99, 6243–6249 CrossRef CAS.
- R. F. Heck and D. S. Breslow, J. Am. Chem. Soc., 1961, 83, 4023–4027 CrossRef.
- G. Natta, R. Ercoli, S. Castellano and F. H. Barbieri, J. Am. Chem. Soc., 1954, 76, 4049–4050 CrossRef CAS.
- P. Wermer, B. S. Ault and M. Orchin, J. Organomet. Chem., 1978, 162, 189–194 CrossRef CAS.
- R. W. Wegman and T. L. Brown, J. Am. Chem. Soc., 1980, 102, 2494–2495 CrossRef CAS.
- F. Ungváry and L. Markó, J. Organomet. Chem., 1980, 193, 383–387 CrossRef.
- E. Folga and T. Ziegler, J. Am. Chem. Soc., 1993, 115, 5169–5176 CrossRef CAS.
- R. J. Klingler and J. W. Rathke, Inorg. Chem., 1992, 31, 804–808 CrossRef CAS.
- R. Tannenbaum, U. K. Dietler, G. Bor and F. Ungváry, J. Organomet. Chem., 1998, 570, 39–47 CrossRef CAS.
- T. A. Barckholtz and B. E. Bursten, J. Organomet. Chem., 2000, 596, 212–220 CrossRef CAS.
- J. W. Rathke, R. J. Klingler and T. R. Krause, Organometallics, 1992, 11, 585–588 CrossRef CAS.
- R. J. Klingler and J. W. Rathke, J. Am. Chem. Soc., 1994, 116, 4772–4785 CrossRef CAS.
- T. Ziegler, L. Cavallo and A. Bérces, Organometallics, 1993, 12, 3586–3593 CrossRef CAS.
- C.-F. Huo, Y.-W. Li, G.-S. Wu, M. Beller and H. Jiao, J. Phys. Chem. A, 2002, 106, 12161–12169 CrossRef CAS.
- S. K. Goh and D. S. Marynick, Organometallics, 2002, 21, 2262–2267 CrossRef CAS.
- C.-F. Huo, Y.-W. Li, M. Beller and H. Jiao, Organometallics, 2003, 22, 4665–4677 CrossRef CAS.
- C.-F. Huo, Y.-W. Li, M. Beller and H. Jiao, Organometallics, 2004, 23, 765–773 CrossRef CAS.
- C.-F. Huo, Y.-W. Li, M. Beller and H. Jiao, Organometallics, 2005, 24, 3634–3643 CrossRef CAS.
- C.-F. Huo, Y.-W. Li, M. Beller and H. Jiao, Chem.–Eur. J., 2005, 11, 889–902 CrossRef CAS PubMed.
- L. E. Rush, P. G. Pringle and J. N. Harvey, Angew. Chem., Int. Ed., 2014, 53, 8672–8676 CrossRef CAS PubMed.
- K. J. Laidler, Chemical Kinetics, Harper & Row, New York, 3rd edn, 1987 Search PubMed.
- S. Maeda and K. Morokuma, J. Chem. Theory Comput., 2011, 7, 2335–2345 CrossRef CAS.
- S. Maeda, T. Taketsugu and K. Morokuma, J. Comput. Chem., 2014, 35, 166–173 CrossRef CAS PubMed.
- S. Maeda and K. Morokuma, J. Chem. Theory Comput., 2012, 8, 380–385 CrossRef CAS.
- G. Liu, R. Volken and M. Garland, Organometallics, 1999, 18, 3429–3436 CrossRef CAS.
- J. A. Osborn, G. Wilkinson and J. F. Young, Chem. Commun., 1965, 17 RSC.
- D. Evans, J. A. Osborn and G. Wilkinson, J. Chem. Soc. A, 1968, 3133–3142 RSC.
- G. Yagupsky, C. K. Brown and G. Wilkinson, J. Chem. Soc. A, 1970, 1392–1401 RSC.
- C. K. Brown and G. Wilkinson, J. Chem. Soc. A, 1970, 2753–2764 RSC.
- C. W. Kohlpaintner, R. W. Fischer and B. Cornils, Appl. Catal., A, 2001, 221, 219–225 CrossRef CAS.
- W. A. Herrmann and C. W. Kohlpaintner, Angew. Chem., Int. Ed. Engl., 1993, 22, 1524–1544 CrossRef.
- R. Schmid, W. A. Herrmann and G. Frenking, Organometallics, 1997, 16, 701–708 CrossRef CAS.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- R. V. Kastrup, J. S. Merola and A. A. Oswald, Adv. Chem. Ser., 1982, 196, 43–64 CrossRef CAS PubMed.
- D. Gleich and J. Hutter, Chem.–Eur. J., 2004, 10, 2435–2444 CrossRef CAS PubMed.
- N. Koga, S. Q. Jin and K. Morokuma, J. Am. Chem. Soc., 1988, 110, 3417–3425 CrossRef CAS.
- T. Matsubara, N. Koga, Y. Ding, D. G. Musaev and K. Morokuma, Organometallics, 1997, 16, 1065–1078 CrossRef CAS.
- S. A. Decker and T. R. Cundari, Organometallics, 2001, 20, 2827–2841 CrossRef CAS.
- A. Grigoropoulos, D. Maganas, D. Symeonidis, P. Giastas, A. R. Cowley, P. Kyritsis and G. Pneumatikakis, Eur. J. Inorg. Chem., 2013, 1170–1183 CrossRef CAS.
- S. Schmidt, G. Abkai, T. Rosendahl, F. Rominger and P. Hofmann, Organometallics, 2013, 32, 1044–1052 CrossRef CAS.
- (a) W. J. Stevens, H. Basch and M. Krauss, J. Chem. Phys., 1984, 81, 6026–6033 CrossRef PubMed; (b) W. J. Stevens, M. Krauss, H. Basch and P. G. Jasien, Can. J. Chem., 1992, 70, 612–630 CrossRef CAS; (c) T. R. Cundari and W. J. Stevens, J. Chem. Phys., 1993, 98, 5555–5565 CrossRef CAS PubMed.
- R. Car and M. Parrinello, Phys. Rev. Lett., 1985, 55, 2471–2474 CrossRef CAS.
- S. Dapprich, I. Komromi, K. S. Byun, K. Morokuma and M. J. Frisch, J. Mol. Struct.: THEOCHEM, 1999, 462, 1–21 CrossRef.
- S. A. Decker and T. R. Cundari, New J. Chem., 2002, 26, 129–135 RSC.
- D. Gleich, R. Schmid and W. A. Herrmann, Organometallics, 1998, 17, 2141–2143 CrossRef CAS.
- D. Gleich, R. Schmid and W. A. Herrmann, Organometallics, 1998, 17, 4828–4834 CrossRef CAS.
- D. Gleich and W. A. Herrmann, Organometallics, 1999, 18, 4354–4361 CrossRef CAS.
- W. R. Rocha and W. B. de Almeida, Int. J. Quantum Chem., 2000, 78, 42–51 CrossRef CAS.
- R. P. Dias and W. R. Rocha, Int. J. Quantum Chem., 2008, 108, 2358–2373 CrossRef CAS.
- R. P. Dias, M. S. L. Prates, W. B. De Almeida and W. R. Rocha, Int. J. Quantum Chem., 2011, 111, 1280–1292 CrossRef CAS.
- C. A. Tolman, Chem. Rev., 1977, 77, 313–348 CrossRef CAS.
- N. C. Handy and A. J. Cohen, Mol. Phys., 2001, 99, 403–412 CrossRef CAS.
- M. Sparta, K. J. Børve and V. R. Jensen, J. Am. Chem. Soc., 2007, 129, 8487–8499 CrossRef CAS PubMed.
- G. Alagona, C. Ghio, R. Lazzaroni and R. Settambolo, Organometallics, 2001, 20, 5394–5404 CrossRef CAS.
- R. Lazzaroni, R. Settambolo, G. Alagona and C. Ghio, Coord. Chem. Rev., 2010, 254, 696–706 CrossRef CAS PubMed.
- R. Lazzaroni, G. Uccello-Barretta and M. Benetti, Organometallics, 1989, 8, 2323–2327 CrossRef CAS.
- A. Raffaelli, S. Pucci, R. Settambolo, G. Uccello-Barretta and R. Lazzaroni, Organometallics, 1991, 10, 3892–3898 CrossRef CAS.
- R. Lazzaroni, R. Settambolo and G. Uccello-Barretta, Organometallics, 1995, 14, 4644–4650 CrossRef CAS.
- R. Lazzaroni, G. Uccello-Barretta, S. Scamuzzi, R. Settambolo and A. Caiazzo, Organometallics, 1996, 15, 4657–4659 CrossRef CAS.
- C. P. Casey, S. C. Martins and M. A. Fagan, J. Am. Chem. Soc., 2004, 126, 5585–5592 CrossRef CAS PubMed.
- G. Alagona, R. Lazzaroni and C. Ghio, J. Mol. Model., 2011, 17, 2275–2284 CrossRef CAS PubMed.
- G. Alagona and C. Ghio, Theor. Chem. Acc., 2012, 131, 1142–1161 CrossRef PubMed.
- G. Alagona and C. Ghio, J. Organomet. Chem., 2005, 690, 2339–2350 CrossRef CAS PubMed.
- G. Alagona, C. Ghio, R. Lazzaroni and R. Settambolo, Inorg. Chim. Acta, 2004, 357, 2980–2988 CrossRef CAS PubMed.
- G. Alagona, C. Ghio and S. Rocchiccioli, J. Mol. Model., 2007, 13, 823–837 CrossRef CAS PubMed.
- G. Alagona, R. Lazzaroni, S. Rocchiccioli and R. Settambolo, Lett. Org. Chem., 2006, 3, 10–12 CrossRef.
- X. Luo, D. Tang and M. Li, Int. J. Quantum Chem., 2006, 106, 1844–1852 CrossRef CAS.
- X. Luo, D. Tang and M. Li, Int. J. Quantum Chem., 2005, 105, 108–123 CrossRef CAS.
- X. Luo, D. Tang and M. Li, J. Mol. Struct.: THEOCHEM, 2005, 714, 61–72 CrossRef CAS PubMed.
- X. Luo, D. Tang and M. Li, J. Mol. Struct.: THEOCHEM, 2005, 714, 179–188 CrossRef CAS PubMed.
- X. Luo, D. Tang and M. Li, J. Mol. Struct.: THEOCHEM, 2006, 763, 75–81 CrossRef CAS PubMed.
- J. J. Carbó, F. Maseras, C. Bo and P. W. N. M. van Leeuwen, J. Am. Chem. Soc., 2001, 123, 7630–7637 CrossRef PubMed.
- F. Maseras and K. Morokuma, J. Comput. Chem., 1995, 16, 1170–1179 CrossRef CAS.
- N. L. Allinger, Y. H. Yuh and J. H. Lii, J. Am. Chem. Soc., 1989, 111, 8551–8566 CrossRef CAS.
- E. Zuidema, L. Escorihuela, T. Eichelsheim, J. J. Carbó, C. Bo, P. C. J. Kamer and P. W. N. M. van Leeuwen, Chem.–Eur. J., 2008, 14, 1843–1853 CrossRef CAS PubMed.
- A. K. Rappe, C. J. Casewit, K. S. Colwell, W. A. Goddard and W. M. Skiff, J. Am. Chem. Soc., 1992, 114, 10024–10035 CrossRef CAS.
- L. A. van der Veen, P. H. Keeven, G. C. Schoemaker, J. N. H. Reek, P. C. J. Kamer, P. W. N. M. van Leeuwen, M. Lutz and A. L. Spek, Organometallics, 2000, 19, 872–883 CrossRef CAS.
- C. R. Landis and J. Uddin, J. Chem. Soc., Dalton Trans., 2002, 729–742 RSC.
- P. W. N. M. van Leeuwen, C. P. Casey and G. T. Whiteker, in Rhodium Catalyzed Hydroformylation, ed. P. W. N. M. van Leeuwen and C. Claver, Kluwer Academic Publishers, Dordrecht, Netherlands, 2000, pp. 63–106 Search PubMed.
- M. Clark, R. D. Cramer and N. Van Opdenbosch, J. Comput. Chem., 1989, 10, 982–1012 CrossRef CAS.
- J. J. Carbó, A. Lledós, D. Vogt and C. Bo, Chem.–Eur. J., 2006, 12, 1457–1467 CrossRef PubMed.
- S. Aguado-Ullate, S. Saureu, L. Guasch and J. J. Carbó, Chem.–Eur. J., 2012, 18, 995–1005 CrossRef CAS PubMed.
- A. L. Watkins and C. R. Landis, J. Am. Chem. Soc., 2000, 132, 10306–10317 CrossRef PubMed.
- S. Aguado-Ullate, L. Guasch, M. Urbano-Cuadrado, C. Bo and J. J. Carbó, Catal. Sci. Technol., 2012, 2, 1694–1704 CAS.
- S. Aguado-Ullate, J. A. Baker, V. Gonzalez-Gonzalez, C. Muller, J. D. Hirst and J. J. Carbo, Catal. Sci. Technol., 2014, 4, 979–987 CAS.
- B. Breit and W. Seiche, J. Am. Chem. Soc., 2003, 125, 6608–6609 CrossRef CAS PubMed.
- U. Gellrich, W. Seiche, M. Keller and B. Breit, Angew. Chem., Int. Ed., 2012, 51, 11033–11038 CrossRef CAS PubMed.
- U. Gellrich, D. Himmel, M. Meuwly and B. Breit, Chem.–Eur. J., 2013, 19, 16272–16281 CrossRef CAS PubMed.
- I. A. Tonks, R. D. Froese and C. R. Landis, ACS Catal., 2013, 3, 2905–2909 CrossRef CAS.
- S. Schmidt, P. Deglmann and P. Hofmann, ACS Catal., 2014, 4, 3593–3604 CrossRef CAS.
- D. Tang, S. Qin, Z. Su and C. Hu, Organometallics, 2007, 26, 33–47 CrossRef CAS.
- K. J. Coutinho, R. S. Dickson, G. D. Fallon, W. Roy Jackson, T. De Simone, B. W. Skelton and A. H. White, J. Chem. Soc., Dalton Trans., 1997, 3193–3200 RSC.
- C. Y. Hsu and M. Orchin, J. Am. Chem. Soc., 1975, 97, 3553 CrossRef CAS.
- I. Schwager and J. Knifton, J. Catal., 1976, 45, 256–267 CrossRef CAS.
- G. Consiglio and P. Pino, Helv. Chim. Acta, 1976, 59, 642–645 CrossRef CAS.
- Y. Kawabata, T. Hayashi and I. Ogata, J. Chem. Soc., Chem. Commun., 1979, 10, 462–463 RSC.
- T. Hayashi, Y. Kawabata, T. Isoyama and I. Ogata, Bull. Chem. Soc. Jpn., 1981, 54, 3438–3446 CrossRef CAS.
- G. Consiglio, P. Pino, L. I. Flowers and C. U. Pittmann Jr, J. Chem. Soc., Chem. Commun., 1983, 612–613 RSC.
- P. Haelg, G. Consiglio and P. Pino, J. Organomet. Chem., 1985, 296, 281–290 CrossRef CAS.
- L. Kollár, G. Consiglio and P. Pino, J. Organomet. Chem., 1987, 330, 305–314 CrossRef.
- L. Kollár, J. Bakos, I. Tóth and B. Heil, J. Organomet. Chem., 1989, 370, 257–261 CrossRef.
- G. Parrinello and J. K. Stille, J. Am. Chem. Soc., 1987, 109, 7122–7127 CrossRef CAS.
- J. K. Stille, H. Su, P. Brechot, G. Parrinello and L. S. Hegedus, Organometallics, 1991, 10, 1183–1189 CrossRef CAS.
- L. Kollár, T. Kégl and J. Bakos, J. Organomet. Chem., 1993, 453, 155–158 CrossRef.
- M. Gomez, G. Muller, D. Sainz, J. Sales and X. Solans, Organometallics, 1991, 10, 4036–4045 CrossRef CAS.
- W. R. Rocha and W. B. de Almeida, Int. J. Quantum Chem., 1997, 65, 643–650 CrossRef CAS.
- J. B. Foresman, T. A. Keith, K. B. Wiberg, J. Snoonian and M. J. Frisch, J. Phys. Chem., 1996, 100, 16098–16104 CrossRef CAS.
- J. Chatt and R. G. Wilkins, J. Chem. Soc., 1952, 273–278 RSC.
- W. R. Rocha and W. B. de Almeida, J. Braz. Chem. Soc., 2000, 11, 112–120 CrossRef CAS PubMed.
- P. Illner, A. Zahl, R. Puchta, N. van Eikema Hommes, P. Wasserscheid and R. van Eldik, J. Organomet. Chem., 2005, 690, 3567–3576 CrossRef CAS PubMed.
- I. Tóth, T. Kégl, C. J. Elsevier and L. Kollár, Inorg. Chem., 1994, 33, 5708–5712 CrossRef.
- B. B. Coussens, F. Buda, H. Oevering and R. J. Meier, Organometallics, 1998, 17, 795–801 CrossRef CAS.
- S. Creve, H. Oevering and B. B. Coussens, Organometallics, 1999, 18, 1967–1978 CrossRef CAS.
- L. Jánosi, T. Kégl and L. Kollár, J. Organomet. Chem., 2008, 693, 1127–1135 CrossRef PubMed.
- P. Pongrácz, T. Papp, L. Kollár and T. Kégl, Organometallics, 2014, 33, 1389–1396 CrossRef.
- L. Kollár, J. Bakos, I. Tóth and B. Heil, J. Organomet. Chem., 1988, 350, 277–284 CrossRef.
- T. Papp, L. Kollár and T. Kégl, Chem. Phys. Lett., 2013, 588, 51–56 CrossRef CAS PubMed.
- W. R. Rocha and W. B. de Almeida, Organometallics, 1998, 17, 1961–1967 CrossRef CAS.
- W. R. Rocha and W. B. de Almeida, J. Comput. Chem., 2000, 21, 668–674 CrossRef CAS.
- C. Gonzalez and H. B. Schlegel, J. Chem. Phys., 1989, 90, 2154–2161 CrossRef CAS PubMed.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899–926 CrossRef CAS.
- W. R. Rocha, J. Mol. Struct.: THEOCHEM, 2004, 17, 133–143 CrossRef PubMed.
- S. Dapprich and G. Frenking, J. Phys. Chem., 1995, 99, 9352–9362 CrossRef CAS.
- J. C. S. da Silva, R. P. Dias, W. B. de Almeida and W. R. Rocha, J. Comput. Chem., 2010, 31, 1986–2000 CAS.
- A. Bedekovits, L. Kollár and T. Kégl, Inorg. Chim. Acta, 2010, 363, 2029–2045 CrossRef CAS PubMed.
- A. E. Reed, R. B. Weinstock and F. Weinhold, J. Chem. Phys., 1985, 83, 735–746 CrossRef CAS PubMed.
- F. W. Bader, Atoms in Molecules–A Quantum Theory, Oxford University Press, Oxford, 1990 Search PubMed.
- R. P. Dias and W. R. Rocha, Organometallics, 2011, 30, 4257–4268 CrossRef CAS.
- T. Papp, L. Kollár and T. Kégl, Organometallics, 2013, 32, 3640–3650 CrossRef CAS.
- M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS PubMed.
- G. Consiglio, F. Morandini, M. Scalone and P. Pino, J. Organomet. Chem., 1985, 279, 193–202 CrossRef CAS.
- Y. V. Babin, A. V. Gavrikov and Y. A. Ustynyuk, Mendeleev Commun., 2008, 18, 12–13 CrossRef CAS PubMed.
- (a) G. Süss-Fink, J. Organomet. Chem., 1980, 193, C20–C22 CrossRef; (b) G. Süss-Fink and G. F. Schmid, J. Mol. Catal., 1987, 42, 361–366 CrossRef.
- J. F. Knifton, J. Mol. Catal., 1988, 47, 99–116 CrossRef CAS.
- L. Alavila and T. A. Pakkanen, J. Mol. Catal., 1993, 84, 145–156 CrossRef.
- M. A. Moreno, M. Haukka, A. Turunen and T. A. Pakkanen, J. Mol. Catal. A: Chem., 2005, 240, 7–15 CrossRef CAS PubMed.
- (a) P. Fong, A. J. Lough, R. H. Morris, A. Mezzetti, E. Rocchini and P. Rigo, J. Chem. Soc., Dalton Trans., 1998, 2111–2114 RSC; (b) J. K. Law, H. Mellows and D. M. Heinekey, J. Am. Chem. Soc., 2002, 124, 1024–1030 CrossRef CAS PubMed; (c) K. Almeida-Leñero, M. Kranenburg, Y. Guari, P. C. J. Kamer, P. W. N. M. van Leeuwen, S. Sabo-Etienne and B. Chaudret, Inorg. Chem., 2003, 42, 2859–2866 CrossRef PubMed.
- M. A. Moreno, M. Haukka, M. Kallinen and T. A. Pakkanen, Appl. Organomet. Chem., 2006, 20, 51–69 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2015 |