
Selective thioacylation of amines in water: a convenient preparation of secondary thioamides and thiazolines†
Abstract
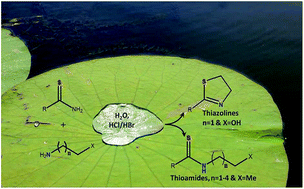
Primary thioamides have been utilised directly in water, without any derivatisation, to selectively thioacylate primary amines. By employing 2-hydroxyethylamines, the reaction can be extended to the preparation of 2-thiazolines via formation of β-hydroxythioamides.
 Please wait while we load your content...
Please wait while we load your content...

