
Bismuth oxide nanoparticles as a nanoscale guide to form a silver–polydopamine hybrid electrocatalyst with enhanced activity and stability for the oxygen reduction reaction†
Settu Murali,
Jen-Lin Chang and
Jyh-Myng Zen*
Department of Chemistry, National Chung Hsing University, Taichung 40227, Taiwan. E-mail: jmzen@dragon.nchu.edu.tw; Fax: +886 4 22850864; Fax: +886 4 22854007; Tel: +886 4 22850864 Tel: +886 4 22854007
First published on 5th December 2014
Abstract
Highly dispersed Ag nanoparticles (Ag NPs) were successfully synthesized on functionalized polydopamine (PDA)@Bi2O3 NPs for use as an electrocatalyst. In the proposed method, a uniform layer of PDA was first coated on Bi2O3 NPs. The surface of the PDA@Bi2O3 can then be used as a nanoscale guide to deposit Ag NPs and hence for the formation of Ag–PDA@Bi2O3 hybrid nanocatalysts. It was found that Ag NPs enhanced the electrocatalytic ability on PDA@Bi2O3 by a synergetic effect for direct 4e− transfer in the oxygen reduction reaction (ORR) with a low overpotential. The surface morphology and lattice fringes of Ag NPs of crystalline nature of the obtained Ag–PDA@Bi2O3 hybrid nanocatalysts were examined through HR-TEM and SAED patterns. The material's purity and chemical functional groups were identified by FT-IR analysis. This strategy provides new opportunities to design and optimize heterogeneous nanocatalysts with tailored size, morphology, chemical configuration and supporting substrates for metal-catalyzed reactions.
Introduction
We report here the application of polydopamine (PDA) coating-directed substrate-immobilization of Ag nanoparticles (Ag NPs) as a promising material in the oxygen reduction reaction (ORR). Note that ORR is essential in various applications, such as fuel cells, metal–air batteries, catalysis, enzymatic reactions and estimation of dissolved oxygen in bio/environment samples.1–6 Recent efforts in ORR electrocatalysis have focused on improving the catalytic activity of Pt alloys, minimizing Pt content by utilizing a core–shell structure, preparing Pt supporting material via synergetic effect and replacing Pt with less expensive materials.1,7,8 For example, several materials, such as Co,1,9 metal oxides/composites,1,3,4 Ag,10–18 and N, S, B, P-doped carbon based materials,19–25 were reported to replace Pt for ORR.Since PDA layer can be formed easily on the surface of Bi2O3 NPs, our strategy is to use the PDA@Bi2O3 as a nanoscale guide to form uniform Ag NPs on the surface of PDA@Bi2O3. Silver can be regarded as a less-expensive and relatively abundant metal, exhibits good ORR activity in alkaline media,15,16 especially for HO2− disproportionation.17,18 In addition, N-containing compounds and polymers were reported to prepare ORR catalyst for tuning the metal nanoparticle size, surface area, complex formation, hetero atom doping with higher stability and durability.1,2,19,20 Synergetic effects of within metal, metal oxide, carbon nano-materials, mixed composites material, etc. were demonstrated to increase the catalytic activity. Of course, the main advantage of PDA is that it could be easily self-polymerized in basic pH medium on any matrix surfaces with multiple applications in variety of fields.26 It has shown good adhesive and reducing properties with higher amount of nanoparticle loading and different structure nanomaterial formation.27 Meanwhile, both PDA-based materials and bismuth-combined composites have also shown enhanced activity in ORR.9,28–33
Herein, an easily modified large surface area Non Hazardous Nano Composite (NHNC) material based on Ag–PDA@Bi2O3 was demonstrated to have enhanced catalytic behavior towards ORR. A key issue for ORR activity at Ag or other catalysts is whether O2 is reduced by two electrons to H2O2 (or HO2−) or four electrons to water (or OH−), i.e., with an n-value of 2 or 4. In general, an n-value of 4 is the preferred pathway, because of both the higher currents available and the unwanted chemical reactivity of H2O2 and its decomposition products toward various fuel cell components. In this paper, we thoroughly studied the electrocatalytic ability of the Ag–PDA@Bi2O3 for direct 4e− transfer in ORR with a low overpotential. The surface morphology, lattice fringes and crystal plane of the obtained Ag–PDA@Bi2O3 hybrid nanocatalysts were examined through HR-TEM and SAED pattern.
Experimental
Dopamine hydrochloride was received from Sigma Aldrich. Sodium dihydrogen phosphate, disodium hydrogen phosphates were purchased from SHOWA chemicals, Japan. Silver nitrate (AgNO3), Tris–HCl, nano MnO2 and bismuth oxide were of ACS grade chemicals. The disposable Screen Printed Carbon Electrode (SPCE) (geometrical area 0.2 cm2) and Ring Disk SPCE (RDSPCE) were purchased from Zensor R&D laboratory, Taichung, Taiwan. Millipore deionized water (18 MΩ cm−1) was used for throughout this experiments. All chemicals were used without further purifications.To prepare the PDA@Bi2O3 composite, 100 mg Bi2O3 was dispersed in 100 mL of Tris–HCl (10 mM) under ultra-sonication for 10 min. Then, 200 mg dopamine–HCl was introduced into the solution and allowed to continuous stirring for 12 h. The obtained precipitate was centrifuged, washed with distilled water and dried at 60 °C. As to the preparation of Ag–PDA@Bi2O3 composite, 5 mg of PDA@Bi2O3 composite was dispersed into 9 mL of water under ultra-sonication for 5 min and then 1 mL (from 20 mM) of AgNO3 (2 mM) solution added into solution under continuous stirring 2 h. Finally, the obtained precipitate was centrifuged, washed with distilled and followed by drying at 60 °C, which is used for further experiments and characterization (Scheme: Fig. S1†).
The coating solution was prepared by dispersing 2 mg of catalyst (Ag–PDA@Bi2O3) in 1 mL water by ultra-sonication for 5 min. Then, 8 μL of coating solution was drop-coated on SPCE or disk surface of RDSPCE and dried at room temperature. The ring electrode of RDSPCE was modified by 5 μL MnO2 coating solution prepared by adding 5 mg MnO2 in 1 mL water and dried at room temperature. The same procedure was followed to prepare Bi2O3/SPCE, PDA@Bi2O3/SPCE, Ag–PDA/SPCE and Ag–Bi2O3/SPCE.
Functionalized PDA was examined by FT-IR (JASCO FT-IR4100, Japan) in the range from 550 to 4000 cm−1. The morphology particle size and lattice fringes were analyzed through High Resolution Transmission Electron Microscopy (HR-TEM) measurements with Surface Area Diffraction Pattern (SAED) (JEM 2010 instrument operating at an accelerating voltage of 200 kV). Dissolved oxygen meter (EUTECH) was used to quantify the amount of oxygen in O2- and Ar-saturated solution.
Electrochemical experiment was carried out using CHI727e and CHI900 for flow injection analysis work station. Three electrode systems were used for electrochemical work station, catalyst modified electrode as a working electrode, Pt wire as an auxiliary electrode and Ag/AgCl as a reference electrode. Cyclic voltammetry experiment was performed in 0.1 M, pH 7.4 Phosphate Buffer Solution (PBS) at a scan rate of 20 mV s−1. Catalyst-modified glassy carbon rotating disk electrode (GCRDE) was used in O2-saturated PBS with sweep rate 20 mV s−1 in LSV. Flow injection analysis experiment was carried out by our group previously reported RDSPCE cell model.40 An Ar-saturated PBS was used in FIA, flow rate 0.3 mL min−1 and 20 μL of O2 saturated solution injected into the flow for the H2O2 intermediate monitoring.
Results and discussion
Characterization of the Ag–PDA@Bi2O3 nanocomposite
The formation of PDA layer on Bi2O3 surface was first characterized by FT-IR spectrum. As shown in Fig. 1, compared to those of DA and PDA spectrum, the stretching vibration at 3200–3500 cm−1 corresponds to N–H and O–H groups. Whereas, the peak at 1602 and 1497 cm−1 corresponds to indole and indoline aromatic ring center, respectively. Finally, the broad peak ranging from 1240 to 1282 cm−1 comes from the C–O asymmetric stretching vibration and C–OH asymmetric bending vibration. This is in consistent with previous studies for characterization of PDA thin films deposited by autoxidation of DA.34,35 The fact that there are no any other extra peaks observed at PDA and PDA@Bi2O3 also confirmed the purity of the as-prepared materials. Overall, melanin like structure PDA layer was strongly formed on the surface of the Bi2O3 NPs.27 | ||
| Fig. 1 Characterization of DA (a), PDA (b) and PDA@Bi2O3 (c) by FT-IR spectroscopy. | ||
Since we propose to use the Bi2O3 NPs as a nanoscale guide for Ag NPs formation, the surface morphology of the catalyst by HR-TEM can provide a direct evidence for such a purpose. At first, as shown in Fig. 2a a pure nanostructured spherical Bi2O3 NPs was observed. Note that the Bi2O3 particle formation density was varied at different particle with higher density particle showing very dark and sharp edges and lower density particle of grey color (Fig. S2a†). Simply by mixing the Bi2O3 NPs with dopamine solution in Tris–HCl, the polymer matrix was effectively coated on the surface of the Bi2O3 NPs, as confirmed from Fig. 2b. Note that this is similar to the polymer matrix layer formed on Bi2O3 NPs to that of previously reported PDA layer formation on Fe3O4.36 The polymer coated thickness of PDA was measured as 50 nm. As shown in Fig. 2c, in the absence of PDA, no obvious Ag NPs were observed for the Ag–Bi2O3 composites. Yet, small amount of silver layer was found on the edges of Bi2O3 surfaces. Note that it could also be visibly observed as yellow color Bi2O3 solution turned into slightly whitish yellow while synthesis the composite (Fig. 2c and S2b†). In other words, the existence of PDA is essential for the successfully loading of large amount of Ag NPs. To prove this, we purposely prepare an Ag–PDA composite via AgNO3 solution mixed with PDA powder under stirring condition. As reported earlier, Ag cations can be reduced by PDA powder from metal ions to metal nanoparticles.26,27,37 As shown in Fig. 2d, large amount of Ag NPs were indeed formed under the PDA condition.
 | ||
| Fig. 2 HR-TEM images of Bi2O3 (a), PDA@Bi2O3 (b), Ag–Bi2O3 (c) and Ag–PDA (d). | ||
We next improve the preparing method simply by mixing the PDA@Bi2O3 with AgNO3. The surface morphology of the as-prepared Ag–PDA@Bi2O3 composite was further studied by HR-TEM. As shown in Fig. 3 and S3,† there is a layer of mixture of Ag film and Ag NPs on the PDA@Bi2O3 surface. The Ag NPs was found to form on PDA@Bi2O3 surface with spherical shape but in different size. The average particles size of Ag NPs were ∼34 nm (Table S1†).37 Note that higher amount Ag NPs were formed on PDA@Bi2O3 surface due to PDA reducing properties.27,37 The crystal nature of the Ag NPs was studied by SAED image. Fig. 3c and d show the lattice fringes and SAED pattern of Ag NPs in Ag–PDA@Bi2O3 composite. It is clear that {111}, {220}, {200} lattice plane of face center cubic (fcc) crystal nature of Ag NPs were formed at the Ag–PDA@Bi2O3 composite. This is advantageous since the pattern is similar to the Ag NPs added to improve the performance of a zinc oxide nanowire ultraviolet photodetector.38
 | ||
| Fig. 3 HR-TEM images of Ag–PDA@Bi2O3 (a), (b) and lattice fringes of silver in Ag–PDA@Bi2O3 (c) and SAED image for crystal nature characterization of Ag NPs in Ag–PDA@Bi2O3 (d). | ||
Electrocatalytic activity of the Ag–PDA@Bi2O3 composite electrode for ORR
Fig. 4 compares the electrochemical behavior of different composite electrodes towards ORR. As can be seen, in the absence of Ag NPs, both Bi2O3 and PDA@Bi2O3 electrodes revealed very low catalytic behavior for the reduction of O2. On the other hand, both Ag NPs on Ag–PDA and Ag layer on Ag–Bi2O3 modified electrodes exhibited good electrocatalytic behavior at ∼−0.6 V vs. Ag/AgCl.37 As to the Ag–PDA@Bi2O3 composite electrode, a lower reduction potential at −0.38 V with an enhanced catalytic activity in ORR was observed. The observation of a totally different cyclic voltammograms also indicates that there is a synergetic effect among PDA, Ag NPs and Bi2O3 enhance the electrocatalytic ability and hence a lower potential for ORR. Compared to those of Ag layer on Bi2O3 and Ag NPs on PDA@Bi2O3, the Ag NPs on PDA with Bi2O3 supporting substrate improved the catalytic activity with a lower reduction potential for ORR. To elucidate this, the ORR electrocatalytic activity of the Ag–PDA@Bi2O3 was investigated more detail in O2-saturated 0.1 M, pH 7.4 PBS using a rotating disk electrode system to verify whether O2 is reduced with an n-value of 2 or 4. | ||
| Fig. 4 Cyclic voltammograms of different composite-modified SPCE for ORR in atmospheric (dashed line), anaerobic (dotted line) and O2-saturated (solid line) PBS (pH = 7.4), respectively. | ||
We purposely compare different electrochemical behavior of ORR at three representative electrodes of Au–RDE, GC-RDE and Ag–PDA@Bi2O3 modified GC-RDE. It is well known that GC electrode catalyzes the ORR with an n-value of 2 and nano-Au electrode involves two step 2e− transfer for ORR and H2O2 reduction, respectively. As shown in Fig. 5a, at the Au–RDE, two step 2e− transfer was truly observed at ∼−0.37 V for ORR and at −0.7 V for H2O2 reduction. The GC-RDE, on the other hand, only shows a 2e− transfer ORR at −0.7 V. It is interesting that the Ag–PDA@Bi2O3 modified electrode shows a single and sharp ORR peak at −0.38 V. In order to quantitatively evaluate the ORR kinetics (including n-values), analysis of the RDE data was done using the Koutecky–Levich (K–L) formalism as given by:20 1/j = 1/jk + 1/(Bω)1/2, in which B = 0.62nFCoDo2/3/v1/6 where j is the measured current density, jk is the kinetic current density, ω is the rotation rate, n is the overall number of electrons transferred in oxygen reduction, F is the Faraday constant (F = 96![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485 C mol−1), Co is the bulk concentration of O2 (0.25 × 10−3 mol L−1), Do is the diffusion coefficient of O2 in the electrolyte (2.51 × 10−5 cm2 s−1),6 and v is the viscosity of the electrolyte. Active surface area can be calculated by Randles–Sevcik equation (Fig. S5 and Table S2†). Fig. 5b and c show the ORR polarization curve at different rotation rate ranging from 150 to 1000 rpm. The ORR current responses were found to increase with the rotation rate, and this process represented a diffusion control process. Fig. 5d express the K–L plot via the inverse current density (j−1) as a function of inverse of the square root of the rotation rate. According to the K–L equation (Table S2†), the n of ORR was calculated to be 3.69 at −0.38 V and jk value of 18.18 mA cm−2. The corresponding K–L curves at various potentials exhibit good linearity, and the slopes remain almost unchanged over the potential range from −0.38 to −0.78 V (Fig. S6†), suggesting that the electron transfer numbers for oxygen reduction at different potentials are similar. The linearity and parallelism of the plots are considered as an indication of first-order reaction kinetics with respect to the concentration of dissolved O2.20 This value is good compared to previously reported Ag-based ORR catalyst10–13,16 and other non-Pt based catalyst.3,9,19–25,29–31
485 C mol−1), Co is the bulk concentration of O2 (0.25 × 10−3 mol L−1), Do is the diffusion coefficient of O2 in the electrolyte (2.51 × 10−5 cm2 s−1),6 and v is the viscosity of the electrolyte. Active surface area can be calculated by Randles–Sevcik equation (Fig. S5 and Table S2†). Fig. 5b and c show the ORR polarization curve at different rotation rate ranging from 150 to 1000 rpm. The ORR current responses were found to increase with the rotation rate, and this process represented a diffusion control process. Fig. 5d express the K–L plot via the inverse current density (j−1) as a function of inverse of the square root of the rotation rate. According to the K–L equation (Table S2†), the n of ORR was calculated to be 3.69 at −0.38 V and jk value of 18.18 mA cm−2. The corresponding K–L curves at various potentials exhibit good linearity, and the slopes remain almost unchanged over the potential range from −0.38 to −0.78 V (Fig. S6†), suggesting that the electron transfer numbers for oxygen reduction at different potentials are similar. The linearity and parallelism of the plots are considered as an indication of first-order reaction kinetics with respect to the concentration of dissolved O2.20 This value is good compared to previously reported Ag-based ORR catalyst10–13,16 and other non-Pt based catalyst.3,9,19–25,29–31
 | ||
| Fig. 5 (a) Cyclic voltammograms for ORR at different RDEs. RDE voltammetry curve for ORR on GCRDE (b) and Ag–PDA@Bi2O3-modified GCRDE (c) in 0.1 M O2-saturated PBS (pH 7.4) at various rotation rates. (c) RDE diffusion curve of different electrodes (GC at −0.9 V, Ag–PDA@Bi2O3 at −0.38 V). (d) K–L plots. | ||
Instead of using RDE system, our group previously reported a simple way to identify the 4e− transfer using a flow injection system.39,40 Fig. S7† illustrates the instrumental setup and the working principle. Note that MnO2 NPs is a well-known catalyst for H2O2 oxidation. As shown in Fig. 6, by injection 20 μL of O2-sat. solution, the ORR reaction was found to occur at applied potential −0.38 V on the Ag–PDA@Bi2O3 modified disk electrode and only little amount of H2O2 produced from this reaction was monitored at the MnO2 NPs modified ring electrode at oxidation potential of 0.8 V.39 Based on the results, the achievement of 4e− transfer ORR was as high as 98% since only ∼2% H2O2 oxidation was observed. Note that, by calculating from the H2O2 formation percentage, n = 3.92 is a further evidence of 4e− transfer in ORR. Since the Ag–PDA@Bi2O3 possessed good electrocatalytic ability towards ORR, its application for use as a sensor for detecting dissolved oxygen was also evaluated.6,14 Fig. S8† shows voltammetric behavior of dissolved oxygen in Ar-saturated 0.1 M PBS (pH 7.4) and a wide linear range from 0.0841–6.048 mg L−1 with a sensitivity of 2.1 μA mg−1 was achieved. It is thus also suitable for application in environmental analysis. Stability is one of the crucial factors in fuel cell and sensor application. The Ag–PDA@Bi2O3 composite also showed good stability in O2-sat. PBS. After 2500 cycles under a scan rate of 200 mV s−1, the maximum current remained similarly except the onset potential shifted positive slightly. This catalyst shows superior stability without any binding material (like Nafion) to modify the electrode due to the superior PDA adhesion properties. Long-term stability of this composite is also excellent to retain 99% of activity towards ORR after 57 days.
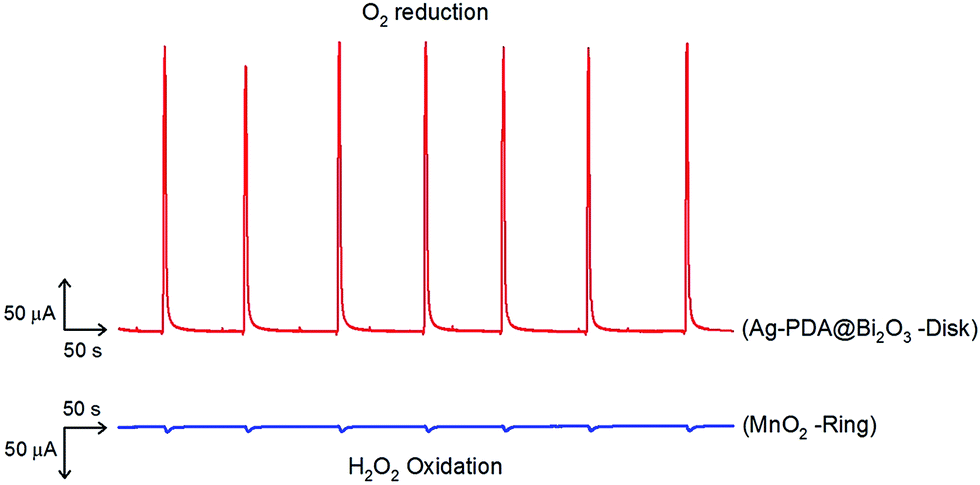 | ||
| Fig. 6 Flow injection analysis of H2O2 oxidation on nano MnO2 catalyst on ring at an operating potential 0.8 V vs. Ag/AgCl and ORR on Ag–PDA@Bi2O3 catalyst coated on disk at detection potential −0.38 V vs. Ag/AgCl with flow rate 0.4 mL min−1. | ||
Conclusions
In summary, we have demonstrated successful synthesis a simple way of Ag–PDA@Bi2O3 nanocomposite with good stability. Face centered cubic lattice crystal nature Ag NPs can be easily formed on the PDA@Bi2O3. The Bi2O3 NPs helped to enhance the catalytic activity of Ag NPs with a lower potential in 4e− transfer pathway of ORR. Electrocatalytic tests for ORR indicated that the synthesized Ag–PDA@Bi2O3 nanocomposite displayed superior activity. The enhanced ORR activity is attributed to the synergistic effects of the composition and dominant face centered cubic lattice crystal nature Ag NPs. This work would be highly impactful in the rational design of future bimetallic alloy nanostructures with high catalytic activity for fuel cell systems.Acknowledgements
The authors gratefully acknowledge financial support from the Ministry of Science and Technology of Taiwan. The author heartedly acknowledge to Prof. Annamalai Senthil Kumar, Environmental and Analytical Chemistry Division, Vellore Institute of Technology University, Vellore, India and Dr Ting-Hao Yang, Department of Soil and Environmental Science, National Chung Hsing University, Taichung, Taiwan for given some useful guidance to carry out this work.References
- S. Guo, S. Zhang and S. Sun, Angew. Chem., Int. Ed., 2013, 52, 8526 CrossRef CAS PubMed.
- K. B. Liew, W. R. W. Daud, M. Ghasemi, J. X. Leong, S. S. Lim and M. Ismail, Int. J. Hydrogen Energy, 2014, 39, 4870 CrossRef PubMed.
- T. H. Yang, S. Venkatesan, C. H. Lien, J. L. Chang and J.-M. Zen, Electrochim. Acta, 2011, 56, 6205 CrossRef CAS PubMed.
- S. Venkatesan, A. S. Kumar, J. F. Lee, T. S. Chan and J. M. Zen, Chem.–Eur. J., 2012, 18, 6147 CrossRef CAS PubMed.
- K. E. Toghill and R. G. Compton, Int. J. Electrochem. Sci., 2010, 5, 1246 CAS.
- C. C. Yang, A. S. Kumar and J. M. Zen, Electroanalysis, 2006, 18(1), 64 CrossRef CAS.
- X. Ma, H. Meng, M. Cai and P. K. Shen, J. Am. Chem. Soc., 2012, 134, 1954 CrossRef CAS PubMed.
- F. Han, T. Zhang, J. Yang, Y. Hu and J. Chen, Adv. Mater., 2014, 26, 2047 CrossRef PubMed.
- H. Wang, X. Bo, A. Wang and L. Guo, Electrochem. Commun., 2013, 36, 75 CrossRef CAS PubMed.
- L. Tammeveski, H. Erikson, A. Sarapuu, J. Kozlova, P. Ritslaid, V. Sammelselg and K. Tammeveski, Electrochem. Commun., 2012, 20, 15 CrossRef CAS PubMed.
- C. L. Lee, H. P. Chiou, C. M. Syu and C. C. Wu, Electrochem. Commun., 2010, 12, 1609 CrossRef CAS PubMed.
- Y. Lu, Y. Wang and W. Chen, J. Power Sources, 2011, 196, 3033 CrossRef CAS PubMed.
- Y. Lu and W. Chen, J. Power Sources, 2012, 197, 107 CrossRef CAS PubMed.
- Y. Zhang, M. Ma and Q. Chen, J. Appl. Electrochem., 2014, 44, 419 CrossRef.
- B. B. Blizanac, P. N. Ross and N. M. Markovic, J. Phys. Chem. B, 2006, 110, 4735 CrossRef CAS PubMed.
- P. Singh and D. A. Buttry, J. Phys. Chem. C, 2012, 116, 10656 CAS.
- D. A. Slanac, W. G. Hardin, K. P. Johnston and K. J. Stevenson, J. Am. Chem. Soc., 2012, 134, 9812 CrossRef CAS PubMed.
- J. Guo, A. Hsu, D. Chu and R. Chen, J. Phys. Chem. C, 2010, 114, 4324 CAS.
- P. Chen, T. Y. Xiao, Y. H. Qian, S. S. Li and S. H. Yu, Adv. Mater., 2013, 25, 3192 CrossRef CAS PubMed.
- R. Liu, D. Wu, X. Feng and K. Mullen, Angew. Chem., 2010, 122, 2619 CrossRef.
- I. Y. Jeon, S. Zhang, L. Zhang, H. J. Choi, J. M. Seo, Z. Xia, L. Dai and J. B. Baek, Adv. Mater., 2013, 25, 6138 CrossRef CAS PubMed.
- Z. Yang, Z. Yao, G. Li, G. Fang, H. Nie, Z. Liu, X. Zhou, X. Chen and S. Huang, ACS Nano, 2012, 6, 205 CrossRef CAS PubMed.
- Z. W. Liu, F. Peng, H. J. Wang, H. Yu, W. X. Zheng and J. Yang, Angew. Chem., Int. Ed., 2011, 50, 3257 CrossRef PubMed.
- L. Yang, S. Jiang, Y. Zhao, L. Zhu, S. Chen, X. Wang, Q. Wu, J. Ma, Y. Ma and Z. Hu, Angew. Chem., Int. Ed., 2011, 50, 7132 CrossRef CAS PubMed.
- C. Zhang, N. Mahmood, H. Yin, F. Liu and Y. Hou, Adv. Mater., 2013, 25(35), 4932 CrossRef CAS PubMed.
- H. Lee, S. M. Dellatore, W. M. Miller and P. B. Messersmith, Science, 2007, 318, 426 CrossRef CAS PubMed.
- Y. Liu, K. Ai and L. Lu, Chem. Rev., 2014, 114, 5057 CrossRef CAS PubMed.
- K. Ai, Y. Liu, C. Ruan, L. Lu and G. Lu, Adv. Mater., 2013, 25, 998 CrossRef CAS PubMed.
- W. Wei, H. Liang, K. Parvez, X. Zhuang, X. Feng and K. Mullen, Angew. Chem., Int. Ed., 2014, 53, 1570 CrossRef CAS PubMed.
- K. E. Lee and B. S. Kim, J. Mater. Chem. A, 2014, 2, 6167 Search PubMed.
- S. M. Sayed and K. Juttner, Electrochim. Acta, 1983, 28, 1635 CrossRef CAS.
- C. Jeyabharathi, J. Mathiyarasu and K. L. N. Phani, J. Appl. Electrochem., 2009, 39, 45 CrossRef CAS.
- I. Arul Raj and K. I. Vasu, J. Appl. Electrochem., 1993, 23, 728 CrossRef.
- R. A. Zangmeister, T. A. Morris and M. J. Tarlov, Langmuir, 2013, 29, 8619 CrossRef CAS PubMed.
- S. Yu, Y. Chen, H. Li, L. Yang, Y. Chen and Y. Yin, J. Mol. Struct., 2010, 982, 152 CrossRef PubMed.
- Y. Xie, B. Yan, H. Xu, J. Chen, Q. Liu, Y. Deng and H. Zeng, ACS Appl. Mater. Interfaces, 2014, 6, 8845 CAS.
- F. Wang, R. Han, G. Liu, H. Chen, T. Ren, H. Yang and Y. Wena, J. Electroanal. Chem., 2013, 706, 102 CrossRef CAS PubMed.
- S. K. Tzeng, M. H. Hon and I. C. Leuc, J. Electrochem. Soc., 2012, 159, H440 CrossRef CAS PubMed.
- A. S. Kumar, S. Sornambikai, S. Venkatesan, J. L. Chang and J. M. Zen, J. Electrochem. Soc., 2012, 159, G137 CrossRef CAS PubMed.
- T. H. Yang, C. Y. Liao, J. L. Chang, C. H. Lien and J. M. Zen, Electroanalysis, 2009, 21, 2390 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Scheme of the work, preparation of Ag–Bi2O3, Ag–PDA, Randles–Sevcik equation for surface area calculation, K–L plot comparison table for n-value calculation, K–L plot diagram of catalyst at different potential, schematic diagram of FIA, oxygen sensor and stability experiment. See DOI: 10.1039/c4ra12989j |
| This journal is © The Royal Society of Chemistry 2015 |