DOI:
10.1039/C4RA12980F
(Paper)
RSC Adv., 2015,
5, 9904-9911
Fabrication of flexible reduced graphene oxide–TiO2 freestanding films for supercapacitor application†
Received
23rd October 2014
, Accepted 7th January 2015
First published on 7th January 2015
Abstract
This paper described the fabrication of a flexible composite film electrode (rGT) composed of reduced graphene oxide (rGO) and TiO2 in an aligned stacking structure of rGO/TiO2 layers using a simple vacuum-assisted filtration method. The SEM images of rGO and rGT films indicate that the rGO film (d = 5.6 ± 0.5 μm) was highly expanded into a layered structure of rGT (d = 7.2 ± 0.5 μm) intercalated by the TiO2 nanoparticles, which prevent the interlayer stacking of graphene sheets. The intensities of C–O and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonding peaks in X-ray photoelectron spectra of rGT films decreased dramatically, as the reduction temperature was increased from 100 to 600 °C, indicating almost complete removal of the oxygenated functional groups. Hence, the electrochemical properties of rGT film electrodes significantly relied on the reduction temperature of graphene oxide (GO)–TiO2 film. The rGT600 electrode (annealed at 600 °C) in 1.0 M Na2SO4 aqueous electrolyte exhibited the specific capacitance of 286 F g−1 in addition to the excellent cycling stability with 93% capacitance retention after 1000 continuous charge/discharge cycles at 1 A g−1. The specific capacitance of the rGT600 electrode in the expanded interlayer stacking structure of rGO and TiO2 particles was by 63% higher than that of the three-dimensional TiO2–graphene hydrogel electrode (175 F g−1, 1 A g−1) prepared using a one-step hydrothermal approach. The modification in the preparation of the composite films comprising rGO sheets and TiO2 particles optimized the supercapacitor electrodes into the expanded interlayer stacking rGO/TiO2 structure, exhibiting excellent electrochemical performance, which is required for the development of advanced micro-size and flexible electrodes in energy storage devices.
O bonding peaks in X-ray photoelectron spectra of rGT films decreased dramatically, as the reduction temperature was increased from 100 to 600 °C, indicating almost complete removal of the oxygenated functional groups. Hence, the electrochemical properties of rGT film electrodes significantly relied on the reduction temperature of graphene oxide (GO)–TiO2 film. The rGT600 electrode (annealed at 600 °C) in 1.0 M Na2SO4 aqueous electrolyte exhibited the specific capacitance of 286 F g−1 in addition to the excellent cycling stability with 93% capacitance retention after 1000 continuous charge/discharge cycles at 1 A g−1. The specific capacitance of the rGT600 electrode in the expanded interlayer stacking structure of rGO and TiO2 particles was by 63% higher than that of the three-dimensional TiO2–graphene hydrogel electrode (175 F g−1, 1 A g−1) prepared using a one-step hydrothermal approach. The modification in the preparation of the composite films comprising rGO sheets and TiO2 particles optimized the supercapacitor electrodes into the expanded interlayer stacking rGO/TiO2 structure, exhibiting excellent electrochemical performance, which is required for the development of advanced micro-size and flexible electrodes in energy storage devices.
1. Introduction
The development of highly efficient energy storage devices has attracted great interest due to the drastic depletion of fossil fuel reserves and the environmental pollution caused by the excessive use of fossil fuels. Furthermore, the invention of small, flexible, and portable electronic devices has triggered the development of high performance micro- and nanoscale power sources.2,3 Among different types of energy storage devices, supercapacitors are attractive systems, which bridge the gap between batteries and conventional capacitors, because of their higher power density and longer life cycle than batteries, as well as higher energy density than conventional capacitors.4,5 Supercapacitors can be classified into two basic types according to their charge storage mechanism: electrical double layer capacitance (EDLC) and pseudocapacitance. The EDLC is generated from the charges that built up electrostatically at the interface of the electrode and electrolyte, whereas the pseudocapacitance is generated from the rapid faradaic reactions that occur on either the surface or bulk of an electrode.5,6 In general, highly porous carbon materials (activated carbon, carbon nanotube, graphene) are used for charge storage via the formation of the EDL at the electrode/electrolyte interface. Almost an unlimited number of charge/discharge cycles can be expected due to the absence of electrochemical degradation in the electrode. Pseudocapacitors that use reactive materials including transition metal oxides and conducting polymers are able to store charges via redox reaction. Pseudocapacitances normally suffer from low power density and poor electrochemical stability due to the irreversible redox processes.6–12 Therefore, current research has been focused mainly on the development of supercapacitor with hybrid electrode containing redox active materials and porous carbon materials for achieving both high power density and energy density.13,14
Graphene, a two-dimensional sheet of sp2-hybridized carbon, is one of the most attractive materials for energy storage devices due to its extraordinary thermal, mechanical, and electrical properties.15 Reduced graphene oxide (rGO), which is generated from graphite oxide, has been a promising route for mass production of chemically modified graphene sheets.16,17 Graphene oxide (GO) obtained via simple and cheap solution processes is hydrophilic due to the presence of various oxygenated functional groups, and forms stable aqueous colloids.15,17,18 GO is electrically insulating material due to disrupted sp2 carbon bonding networks, and, thus, not suitable for supercapacitor applications. Chemical or high temperature treatment of GO is required to convert it to the electrically conductive rGO.19 However, rGO inclined to restack and aggregate due to the strong hydrophobic and π–π interactions between rGO sheets, which lead to reduced specific surface area, thereby resulting in poor capacitive performance.20,21 The restacking of rGO sheets can be suppressed by incorporating secondary materials such as RuO2.5 polyaniline nano fibers,22 and carbon black composites23 for porous composite electrode. Titanium oxide (TiO2) was investigated for the electrode material in energy storage devices due to the pseudocapacitance behavior, excellent chemical stability, eco friendliness, and low production cost.1,24–27 However, the poor electrical conductivity of the TiO2 has limited its use in many electronic applications.28 Incorporation of conductive rGO into TiO2 electrode would overcome the shortcomings in the conductivity and mechanical flexibility of either rGO or TiO2 electrode. Many methods have been proposed to prepare the graphene–TiO2 hybrids composites for supercapacitor electrodes. For example, TiO2 nanoparticles with particle sizes in the range of 8–15 nm were decorated over functionalized graphene by sol–gel methods.29 Xiang and co-workers24 synthesized rGO/TiO2 round-shape nanoparticles and fiber-shape nanobelts using hydrothermal approach and studied the effect of the TiO2 shape on the supercapacitor performance. Additionally, graphene–TiO2 hybrid nanostructure with uniformly distributed TiO2 nanoparticles on the graphene surface was successfully synthesized using microwave-assisted method.27 However, these rGO/TiO2 electrodes required the use of poly(vinylidene fluoride) (PVDF) binder, which severely decreased the electrode conductivity and porosity, leading to poor capacitive performance.
The rapid development of portable and flexible electronic devices has triggered a concerted effort among researchers to develop high performance flexible energy storage devices. Recent studies related to the flexible supercapacitors have shown promising electrochemical performances particularly of those devices composed of carbon-based electrode.30–33 For example, free-standing SWNT/PANi composite film (synthesized using in situ electrochemical polymerization/degradation process)30,31 could show high specific capacitance of 706 F g−1 in a three electrode system. A flexible asymmetric supercapacitor composed of graphene/carbon nanotube/MnO2 (positive electrode) and graphene/carbon nanotube/polypyrrole (negative electrode) operating at a cell voltage of 1.6 V in aqueous electrolyte showed high energy density and power density of 22.8 W h kg−1 and 860 W kg−1, respectively.32 In addition, 3D graphene hydrogel films used in a solid state flexible supercapacitor achieved high specific capacitance of 186 F g−1 at 1 A g−1 in polymer electrolyte.33
Here, we report the fabrication of a binder-free composite freestanding film composed of rGO and TiO2 (rGT) for supercapacitor electrode, whereby TiO2 particles were formed via the hydrolysis of Ti(SO4)2 on GO sheets. The resulting homogenous GO–TiO2 dispersion was filtered using a vacuum-assisted filtration to form GO–TiO2 (GT) film, which was converted to rGT during a thermal treatment at 600 °C. These preparation procedures led to the optimization of the rGT electrode structure with an expanded stacking rGO/TiO2 layers, exhibiting the highly improved electrochemical properties, compared to the one-step hydrothermal approach for three-dimensional (3D) TiO2–graphene hydrogel electrode, which was developed by Zhang and co-workers.1 Simultaneous reduction of GO and crystallization of TiO2 is one of the optimal approaches as it provides good contact between the two materials with defined crystallized morphology.34 The highly expanded layer-structured rGT electrode exhibited the specific capacitance of 286 F g−1 at a current density of 1 A g−1, which was by 63% higher than that of the 3D TiO2–graphene hydrogel electrode (175 F g−1 at 1 A g−1).1
2. Experimental
2.1. Materials
Natural graphite powder (particle size < 20 μm), potassium permanganate (KMnO4), potassium persulfate (K2S2O8), titanium sulfate (Ti(SO4)2) and phosphorus pentoxide (P2O5) were purchased from Sigma-Aldrich, and used as received. Concentrated sulfuric acid (H2SO4) was purchased from Daejung Chemicals (South Korea), and hydrochloric acid (HCl, 35%) and hydrogen peroxide (H2O2, 30%) were obtained from OCI Company Ltd. (Korea). Tin-doped indium oxide (ITO glass, 8 Ω sq−1, d = 100 mm) was purchased from MEMC Electronic Materials Inc.
2.2. Preparation of graphene oxide (GO)
GO solution was synthesized according to the modified Hummers method.35 First, 10 mL of concentrated sulphuric acid (H2SO4) in 500 mL round bottom flask was heated to 80 °C, and then 0.9 g of potassium persulfate (K2S2O8) and 0.9 g phosphorus pentoxide (P2O5) were added into the solution while stirring. After stirring for 30 min, 1 g of graphite powder was added at 80 °C for 4.5 h. Upon being cooled to RT, the mixture was diluted with 250 mL of distilled water, and kept for 12 h at RT. The mixture was then filtrated, washed at least 5 times with distilled water, and dried in vacuum oven. The dried solid sample was added to 40 mL of concentrated sulphuric acid in 500 mL round bottom flask in an ice bath. Then, 5 g of potassium permanganate (KMnO4) was slowly added into the mixture (5 g per 40 minutes) while keeping the temperature under 10 °C. The reaction mixture was heated at 35 °C for 2 h (a color change from black to greenish brown), and followed by dilution with 85 mL of DI water (Caution: the temperature must be kept at <35 °C throughout). After that, 250 mL of DI water and 10 mL of 30% aqueous hydrogen peroxide (H2O2) was added into the mixture. Bubbles were observed from the aqueous mixture along with a color change to brilliant yellow. Then, the mixture was allowed to settle for 12 h; the clear supernatant was removed and the sediment was washed with H2SO4 (5 wt%), H2O2 (0.5 wt%) and HCl (10 wt%) solutions, repeatedly. The dark yellow sediment was washed with DI water. This washing step was repeated, until no layer separation was observed after centrifuging at 4000 rpm. Finally, the synthesized GO solution was dialyzed against DI water for 7 days, and dried in vacuum oven.
2.3. Preparation of reduced graphene oxide/titanium dioxide (rGT) composite films
The GT composite film was prepared based on the previous reports.36,37 To prepare a GT dispersion, 20 mg of dried GO and 60 mg of Ti(SO4)2 were dispersed in 40 mL of DI water, and the mixture solution was stirred and heated at 60 °C for 24 h. In this procedure, TiO2 nanoparticles were formed during the hydrolysis of Ti(SO4)2, and attached to GO sheet. The obtained product was centrifuged, and washed 5 times with distilled water, and then re-dispersed in the mixture of 20 mL of ethanol and 28 mL of DI water, and kept in an oven at 60 °C for 3 days. The composite sample was centrifuged, and washed with DI water at least 5 times to eliminate any impurities. Finally, the obtained GT composite was dispersed in 20 mL of distilled water. GT composite freestanding film was prepared by vacuum filtration of the 20 mL of composite solution through cellulose acetate membrane (50 mm diameter and 0.2 μm pore size), followed by peeling off from the membrane filter after drying, as shown in Fig. 1. The obtained-freestanding GTx (x = 100, 200, 300, 400, 500, and 600 °C) films were thermally reduced at various temperatures ranging from 100 to 600 °C in a tube furnace in an argon atmosphere with heating rate of 10 °C min−1. GO film was also prepared using the same experimental procedure for comparison with the GT film.
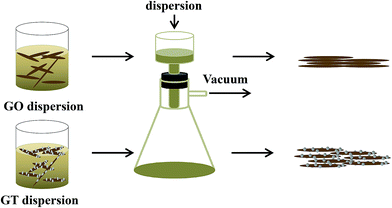 |
| | Fig. 1 Schematic of the formation of GO film (top), and GT (bottom) film. | |
2.4. Material characterization
The cross sectional view and surface morphology of the rGO and rGT films were investigated using field emission scanning electron microscopy (FE-SEM) (JEOL JSM-7001F). The microstructure and grain size of the GO–TiO2 composite were determined using high-resolution transmission electron microscope HRTEM (JEOL JEM 3010) with an accelerating voltage of 300 kV. The structural characteristics were investigated using Raman spectroscopy (Raman-LTPL) at λex = 532 nm, and FT-IR spectroscopy (Nicolet MAGNA 560-FTIR). The electrical conductivity of the samples was measured using the four probe method on CMT-Series (Jandel instrument) at RT, and the elemental compositions of the films were investigated using X-ray photoelectron spectroscopy (XPS), and PHI 5000 VersaprobeII (Ulvac-PHI. Inc.). X-ray diffraction (XRD) pattern of the samples were achieved using Bruker D8 Discover with Cu target of X-ray tube. The wettability of the films was assessed using the face contact-angle meter (Kyowa Interface Science-CA-DT). The water contact angle was measured 5 min after a fixed volume of water droplet was dropped onto the film.
2.5. Electrochemical characterization
All electrochemical measurement of cyclic voltammetry, galvanostatic charge/discharge of rGT films were investigated by scanning over the potential range of 0.0 to 1.0 V in an aqueous 1 M Na2SO4 solution with using potentiostat/galvanostat (Ivium-Stat Technologies compactstat) with the standard three-electrode cell configuration, whereby a platinum wire was used as counter electrode, Ag/AgCl (1 M KCl) was used as reference electrode, and an ITO-coated glass was used as current collector. The electrode with an active area of 0.1 cm2 was being exposed to the electrolyte during electrochemical measurements.
3. Results and discussions
3.1. Preparation of rGO and rGT freestanding films
A stable homogeneous suspension of GO (Fig. 2a, left) was formed in an aqueous solution owing to the hydrophilic oxygenated functional groups. The thermal hydrolysis of Ti(SO4)2 in the GO suspension resulted in the formation of GT precipitates (Fig. 2a, right). Free standing GT and GO films were obtained by filtering the GT and GO suspension through a cellulose acetate filter, respectively. The as-prepared freestanding films of GT and GO were annealed at 600 °C to synthesize rGT600 and rGO600, respectively. The resulting films appeared robust and flexible without formation of cracks upon bending, as shown in Fig. 2b–e.
 |
| | Fig. 2 (a) GO colloidal dispersion (left) and GT composite dispersion (right). Digital images of the freestanding (b) GO, (c) rGO600, (d) GT, and (e) rGT600 films. Insets display the flexibility of the corresponding films upon bending. | |
The surface and cross-sectional morphologies of the rGT600 and rGO600 films were characterized using FESEM, as shown in Fig. 3. The rGT600 film featured rough and crumpled morphology, indicating the formation of TiO2 particles that attached homogeneously onto the surface of rGO600 sheets (Fig. 3a). The rGO600 film featured wrinkled morphology typical of pure graphene sheet (Fig. 3b). The cross-sectional images of rGT600 (Fig. 3c) revealed a layer-by-layer loosely-packed structure with larger interlayer spacing, compared with that of rGO600 film (Fig. 3d). The cross-sectional thickness of rGT600 and rGO600 were measured to be 7.2 (±0.5) μm and 5.6 (±0.5) μm, respectively. The thickness increase clearly indicated the expansion in the interlayer spacing of GT600 sheets due to the TiO2 nanoparticles, which were intercalated into the sheets of rGO sheets. The crumpled structure of rGT600 film would generate enlarged pores accessible to the electrolyte ions, as compared to the compact stacked structure of rGO600 film.38
 |
| | Fig. 3 Surface morphology and cross sectional view of rGT600 film (a and c) and rGO600 film (b and d). | |
3.2. Material characterization of freestanding rGO and rGT films
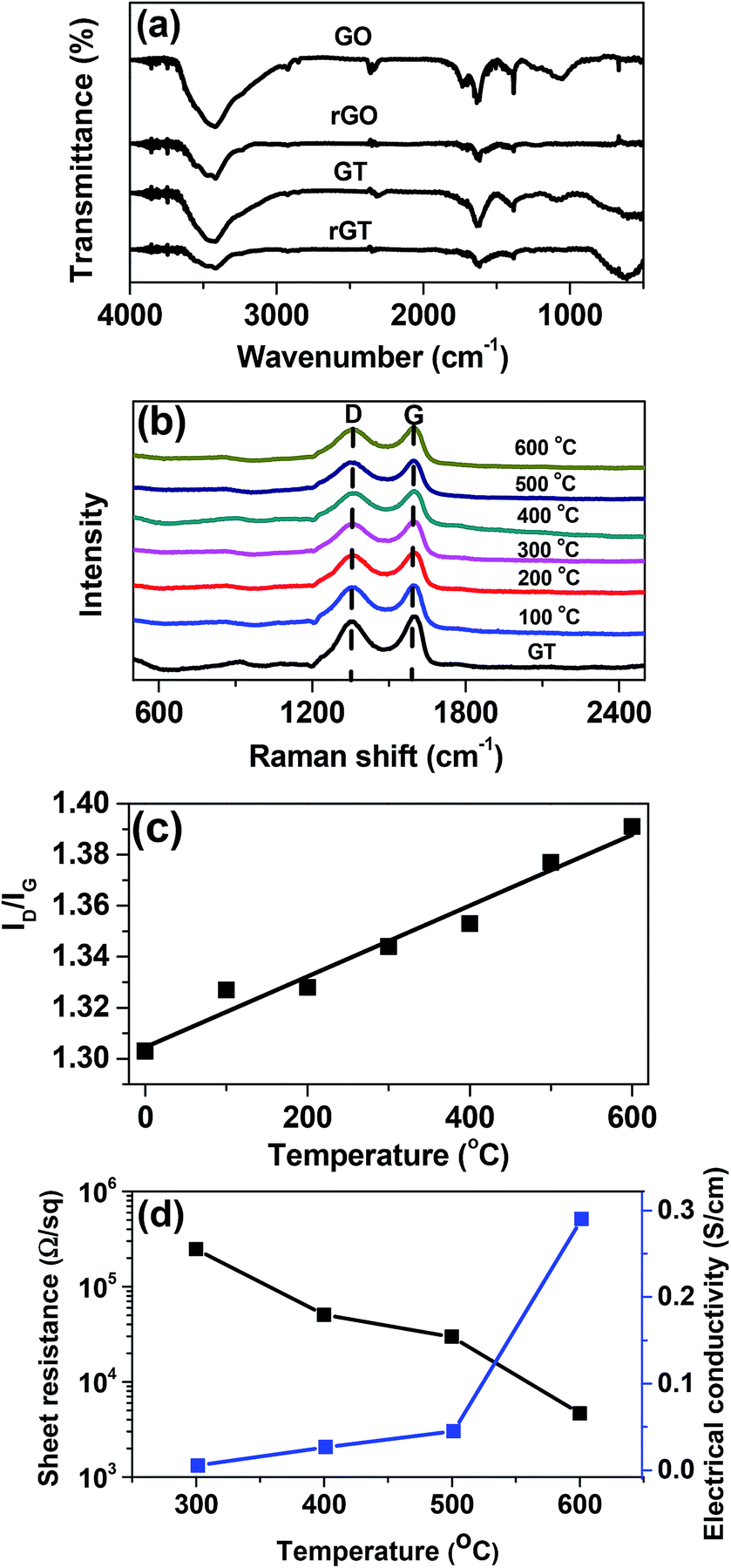
The GT films were converted to rGT films by thermal annealing at different temperature (100–600 °C). The effect of the thermal treatment on the reduction degree of GT was investigated using FTIR spectroscopy, as shown in Fig. 4a. The FTIR spectra of the GT composite films showed a strong absorption peak at 600 cm−1, which was ascribed to Ti–O–Ti vibration of TiO2 particles formed on the GO flakes.27 The broad peaks at 3400, 1380, and 1050 cm−1 were attributed to O–H, C–OH, and C–O stretching vibrations, respectively, indicating the presence of the epoxide and hydoxyl groups on the GO layers.39 The peak at 1740 and 1620 cm−1 were assigned to CO stretching of COOH (carboxyl) functional group and aromatic CC, respectively.40 After thermal annealing at 600 °C, the spectra of rGO and rGT films exhibited drastic decrease in almost all peaks of the oxygenated functional groups, indicating the complete reduction of GO to rGO.
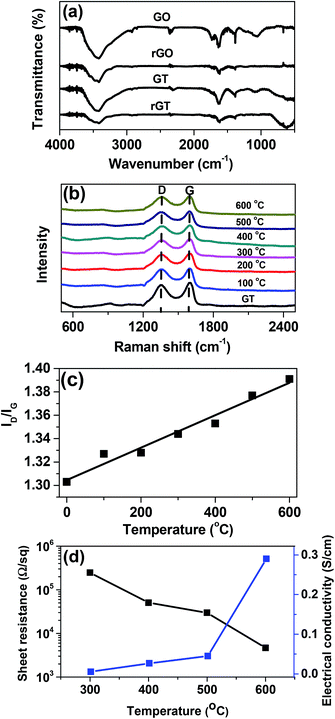 |
| | Fig. 4 (a) IR spectra of GO, rGO600, GT, and rGT600, (b) Raman spectra of GT and rGT films reduced at different temperatures. (c) The plot for the ID/IG value of rGT films vs. reduction temperature. (d) Plot of the sheet resistance and electrical conductivity of GT films as a function of the reduction temperature. Note that the sheet resistance of the GT films was measured using a standard four-point probe method. | |
Fig. 4b showed the Raman spectra of GT before and after thermal treatment at different temperature (100–600 °C). The characteristic D (1340 cm−1) and G (1595 cm−1) bands of GO indicate the presence of disordered sp3 and sp2 carbon atoms, respectively. The intensity of D to G ratio (ID/IG) is widely used to estimate the degree of both the disorder and the crystallite size in graphitic materials.41,42 The ID/IG values of the rGT films increased linearly with increasing annealing temperatures (Fig. 4c), indicating the fact that the oxygenated functional groups were removed more effectively at higher temperature along with increase of the sp2 domains.43 Furthermore, the sheet resistance of the rGT film prepared at different reduction temperature was measured using four-point probe technique, as shown in Fig. 4d. The films annealed at temperature below 200 °C showed insulating properties. However, the sheet resistance of the films decreased by about two orders of magnitude from 2.5 × 105 to 4.7 × 103 Ω sq−1, when the reduction temperature was increased from 300 to 600 °C. The electrical conductivity of the rGT films recalculated from the corresponding sheet resistances increased exponentially from the insulating state to 0.29 (±0.03) S cm−1, indicating great improvement in the electrical conductivity of the films due to the incorporation of the highly conductive rGO component into the composite rGT–TiO2 electrodes.
The reduction of GT to rGT films at different temperature was evaluated based on their elemental compositions that were determined using X-ray photoelectron spectroscopy (XPS), and were compared with those of the pure GO. A high-resolution XPS survey scans of the rGT film annealed at different temperature (Fig. 5a) showed three distinct peaks; O 1s (532 eV), C 1s (285 eV) and Ti 2p (460 eV).27,44,45 These three characteristic peaks were deconvoluted to resolve the individual peaks of the functional groups obtained from the detailed scan. The binding energy (eV) and the relative composition (%) of the functional groups are listed in Table 1 for comparisons. Deconvolution of O 1s peak for GO film (Fig. 5b) exhibited three distinguishable peaks, which were assigned to various oxygenated GO functional groups; OC–O/CO (531.6 eV), C–O (532.4 eV), and H2O (533.8 eV).45 In addition, two additional peaks of Ti–O (529.6 eV) and O22− (530.1 eV) were observed from the rGT film.28,44 A profound reduction in the peak intensity of C–O bonding (532.4 eV) was observed from rGT versus GO films, as the reduction temperature increased to 600 °C, indicating the elimination of oxygenated functional groups including hydroxyl, carbonyl and carboxyl groups.45 Furthermore, deconvolution of C 1s peak (Fig. 5c) showed characteristic peaks of C–C/CC (284.4 eV), C–OH (285.6 eV), C–O (286.5 eV), CO (287.2 eV), and OC–O (288.8 eV) in the GO and rGT films, indicating the presence of oxygenated functional groups on the GO sheet.1,25–27,44 The peak intensity of C–O and CO bonding decreased dramatically, when the reduction temperature was increased to 600 °C, indicating almost complete removal of the oxygenated functional groups including hydroxyl, epoxy, and carbonyl.45 The deconvolution of Ti 2p for Ti atom (Fig. 5d) showed two characteristic peaks corresponding to Ti 2p3/2 (459.4 eV) and Ti 2p1/2 (465.2 eV). The presence of Ti–O peaks in the GT film before and after thermal treatment implied the high thermal stability of the TiO2 particles.25
 |
| | Fig. 5 XPS spectra of GO, rGT200, rGT400, rGT600 films. (a) The survey scan, (b) comparison of O 1s, (c) comparison of C 1s, (d) scaled Ti 2p of rGT600 film. | |
Table 1 The binding energy (eV) and composition (%) of the functional groups comprised in C 1s and O 1s peaks in XPS of the GO and rGT at different reduction temperature
| Samples |
O 1s |
C 1s |
| H2O |
C–O |
OC–O/CO |
O22− |
Ti–O |
OC–O |
CO |
C–O |
C–OH |
C–C |
| GO |
(eV) (%) |
533.8 (6.4) |
532.4 (72.6) |
531.6 (21.0) |
— |
— |
288.5 (7.4) |
287.3 (16.3) |
286.4 (34.5) |
285.6 (5.5) |
284.4 (36.3) |
| rGT200 |
(eV) (%) |
533.7 (13.5) |
532.3 (41.9) |
531.1 (17.5) |
530.1 (3.9) |
529.6 (23.2) |
288.5 (10.3) |
287.3 (12.1) |
286.3 (31.3) |
285.6 (9.5) |
284.3 (36.8) |
| rGT400 |
(eV) (%) |
533.7 (8.9) |
532.4 (23.0) |
531.7 (24.6) |
530.3 (5.3) |
529.9 (38.2) |
288.5 (11.4) |
287.2 (8.3) |
286.4 (14.6) |
285.5 (12.6) |
284.4 (53.1) |
| rGT600 |
(eV) (%) |
533.8 (1.2) |
532.5 (1.4) |
531.6 (25.3) |
530.4 (6.0) |
530.0 (66.1) |
288.5 (12.8) |
287.3 (6.0) |
286.5 (9.6) |
285.6 (13.7) |
284.5 (57.9) |
The morphology and structure of the GO and GT were characterized using HRTEM. A typical wrinkle structure of GO sheet was shown in Fig. S1(a).† TiO2 particles with the average particle size of 5 nm were uniformly deposited on the GO sheet with high density (Fig. S1b†). The lattice fringe of TiO2 nanoparticle (Fig. S1c†) displayed the lattice spacing of the anatase (101) plane of TiO2 to be 0.35 nm, which agreed well with the lattice spacing of TiO2 reported previously.25,46 The crystalline structures of GO and rGT at different temperature were determined by powder X-ray diffraction measurement, as shown in Fig. S2.† The XRD pattern of GO showed a pronounced diffraction peak at 2θ of 12°, which was attributed to (002) plane of GO.47 The diffraction peak of GT and rGT heated at different temperature were observed at 2θ of 25°, 48° and 55°, corresponding to (101), (200), and (105) planes of anatase titania, respectively.25 By increasing the temperature to 600 °C, the GO peak in rGT at 12° disappeared completely and a new broad peak appeared at 2θ of 22°, indicating reduction of GO to rGO.27
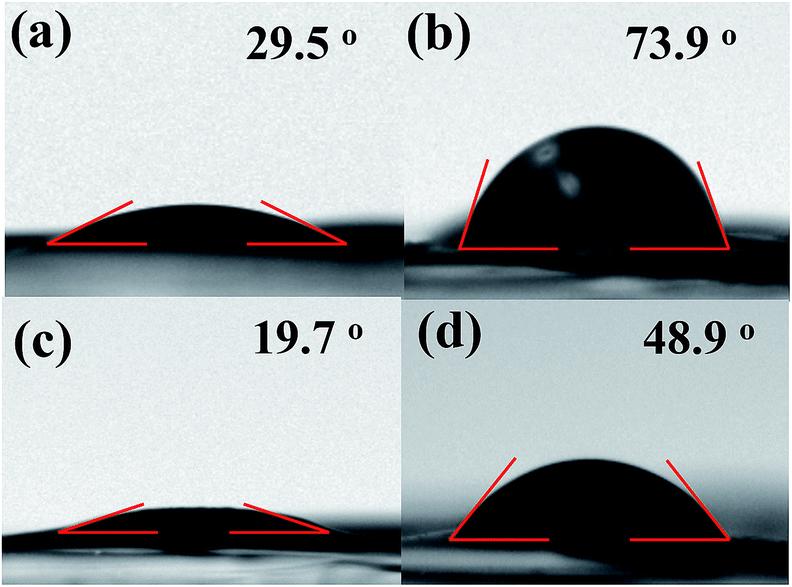
The wettability of an electrode is an additional critical factor to decide the accessibility of aqueous electrolytes into the electrode. Thus, the wettability of rGO600 and rGT600 film surfaces was assessed based on the stationary water contact angle. The water contact angle (29.5°) of GO drastically increased to 73.9° as reduced to rGO600 after thermal reduction at 600 °C, indicating the increased surface hydrophobicity, as shown in Fig. 6. However, the contact angle (19.7°) of GT increased not strongly for rGT600 (48.9°) compared to the case of the GO and rGO600. The enhanced water wettability of both the GT and rGT600 electrodes could be ascribed to the involvement of the hydrophilic component TiO2 in addition to the increased film porosity, which was attributed to the crumpled and TiO2-intercalated structure of the GT films.
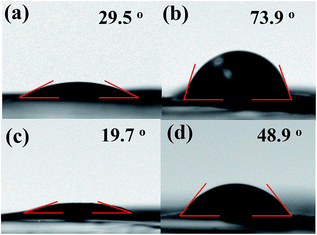 |
| | Fig. 6 Water contact angle of (a) GO, (b) rGO600, (c) GT, and (d) rGT600. | |
3.3. Electrochemical properties of the rGT electrode in a three-electrode setup
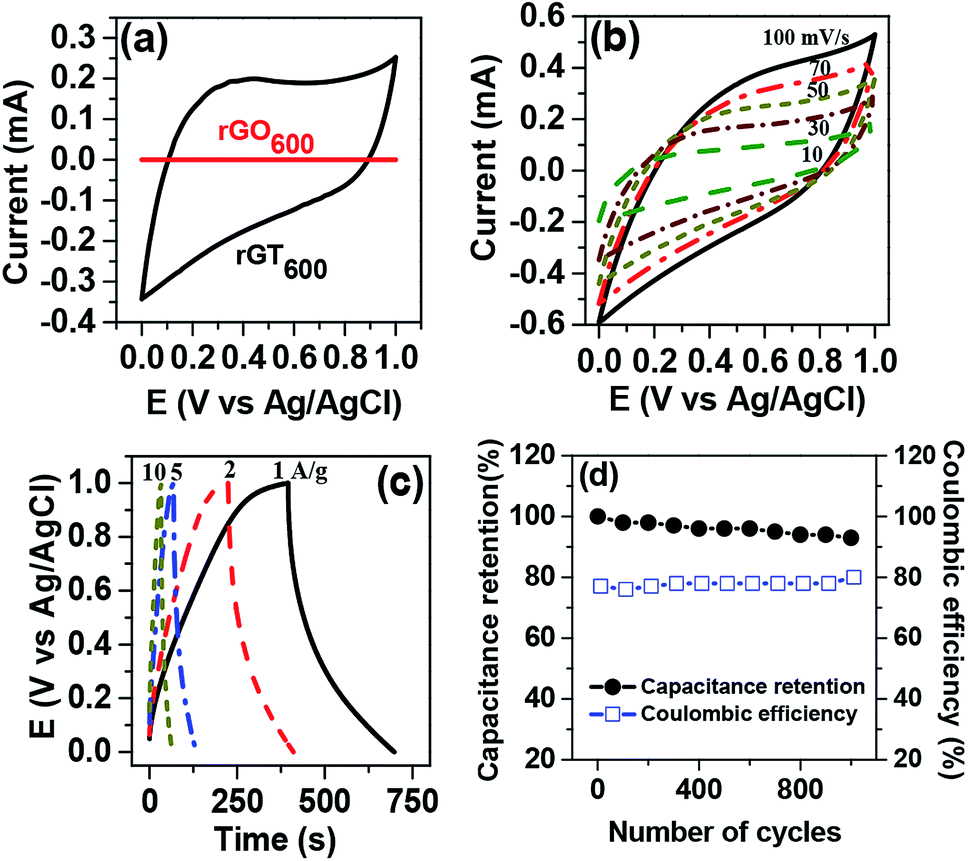
The capacitive performance of the rGTx electrode was evaluated using cyclic voltammetry (CV) and galvanostatic charge/discharge technique in 1 M Na2SO4 aqueous electrolyte. The nearly-rectangular CV curve of rGT600 electrode appeared relatively larger compared to that of the rGO600 electrode, indicating the higher capacitive performance, as shown in Fig. 7a. The improved capacitive performance of the rGT600 electrode versus the rGO600 electrode could be attributed to the improved hydrophilicity of the rGT600, which enhanced the accessibility of the aqueous electrolyte into the rGT600 electrode. The area under the CV curve of the rGT600 electrode (Fig. 7b) increased with the scan rate ranging from 10 to 100 mV s−1. However, the CV curve was slightly distorted due to the increase in the electrode resistance at a high scan rate of 100 mV s−1.48 Fig. 7c showed the charge/discharge curves of the electrode, which was measured at various current densities ranging from 1 to 10 A g−1 within the potential range of 0–1 V. Asymmetrical behavior of the charge/discharge curve was observed for the electrode at low current densities, which can be ascribed to the redox reaction of TiO2.27 However, the capacitive performance of the rGT600 electrode decreased upon increasing the scan rate and the current density, due to the high electrical and ionic resistances of the electrode at high charging/discharging rates. At low scan rates, nearly the entire electrode is accessible to the diffusion-controlled intercalation/deintercalation of electrolyte ions, resulting in the improved specific capacitance of the electrode.49,50
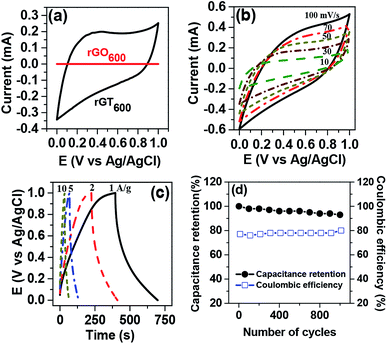 |
| | Fig. 7 (a) Comparison of CV curve for rGO600 and rGT600. (b) CV curve of rGT600 electrodes at various scan rates (c) galvanostatic charge/discharge curves of rGT600 electrodes at various current densities (d) cycling stability and coulombic efficiency of rGT600 electrode. | |
The specific capacitances of rGT electrode were determined from the galvanostatic discharge curve as a function of the current density (Fig. 7c), according to the following eqn (1)48
where
I is the current loaded, Δ
t is the discharge time, Δ
E is the potential range after IR drop during discharge process, and
m is the mass of the working electrode of rGT. The maximum specific capacitance of the rGT electrode annealed at 600 °C was calculated to be 286 F g
−1. The resulted value is much higher, compared to a closely related electrode like TiO
2–graphene nanocomposite hydrogel (175 F g
−1 at 1 A g
−1), which was prepared using hydrothermal approach reported in a previous literature.
1 The improvement of specific capacitance by the rGT was attributed to the loosely-stacked layered structure of the rGT electrode, in which the TiO
2 nanoparticles was formed directly on the graphene sheets,
34 compared to the bulk nanocomposite of TiO
2–graphene hydrogel electrode.
1 The good contact between the two materials can be beneficial for rapid charge transfer by electron injection from TiO
2 into the graphene sheets. Furthermore, the average energy density (
E, W h kg
−1) and power density (
P, W kg
−1) of the rGT
600 electrode were calculated using the following
eqn (2) and
(3):
27where the
Cs is the specific capacitance of the electrode material (F g
−1),
t is the discharge time of the charge/discharge curve, and Δ
V is the potential window. The average energy density and power density of the rGT
600 electrode were calculated to be 39.7 W h kg
−1 and 472 W kg
−1 at 1 A g
−1, respectively. The rGT electrode showed a tendency for the area under CV and the charge/discharge curves to increase upon the reduction temperature for rGT, which increased from 300 to 600 °C, as shown in Fig. S3(a) and (b) in ESI.
† The long-term cycling stability and coulombic efficiency of rGT
600 electrode in 1.0 M Na
2SO
4 aqueous electrolyte (
Fig. 7d) were evaluated using galvanostatic charge/discharge measurement at 1 A g
−1 for 1000 cycles in the potential range of 0 to 1 V. The rGT
600 electrode exhibited excellent cycling stability with 93% capacitance retention after 1000 continuous charge/discharge cycles and a coulombic efficiency of almost 80%.
4. Conclusion
In summary, we have fabricated a highly flexible freestanding film electrode composed of graphene and TiO2 using a simple vacuum-assisted filtration, resulting in the aligned stacking structure of GO/TiO2 layers. The TiO2 intercalated into GO layers, led to the formation of crumpled GO/TiO2 films, in which large pores accessible to electrolytes would be generated. The electrical conductivity of an electrode is a critical factor to decide the level of energy storage in supercapacitor. The conductivity of the hybrid rGT electrode strongly relies on the reduction state of GO, which was investigated as a function of the annealing temperature. The reduction state of GO in the hybrid electrode was evaluated using RAMAN and XPS, as the annealing temperature was ranged from 100 to 600 °C. From RAMAN spectra of the rGT films, the ID/IG values increased linearly with increasing annealing temperatures from 100 to 600 °C, indicating the fact that the oxygenated functional groups were removed more effectively at higher temperature along with increase of the sp2 domains. In addition, the peak intensity of C–O and CO bonding, which was determined from XPS of rGT films, decreased dramatically, as the reduction temperature was increased to 600 °C, indicating almost complete removal of the oxygenated functional groups including hydroxyl, epoxy, and carbonyl. The sheet resistances of the rGT films decreased exponentially by two orders of magnitude from 2.5 × 105 to 4.7 × 103 Ω sq−1, as the reduction temperature of GT (to rGT) increased from 300 to 600 °C. These results indicated that the thermal reduction of the GT at 600 °C was an optimal condition for the resulting film electrode, which is conductive, robust, and flexible. The rGT600 film electrode exhibited the specific capacitance of 286 F g−1 at the 1 A g−1, which is much higher than the closely related electrode, TiO2–graphene nanocomposite hydrogel (175 F g−1 at 1 A g−1), which was prepared using hydrothermal approach reported in a previous literature.1 The long-term cycling stability of rGT600 electrode in 1.0 M Na2SO4 aqueous electrolyte exhibited excellent cycling stability with 93% capacitance retention after 1000 continuous charge/discharge cycles at 1 A g−1 under the potential range of 0 to 1 V. The hybrid rGT600 electrode exhibited the excellent electrochemical properties, which are required for the development of advanced micro-size and flexible electrodes in energy storage devises.
Acknowledgements
This research was supported by the Research Grant of Incheon National University in 2013, and Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (20110015807).
Notes and references
- Z. Zhang, F. Xiao, Y. Guo, S. Wang and Y. Liu, ACS Appl. Mater. Interfaces, 2013, 5, 2227–2233 CAS.
- H. R. Byon, S. W. Lee, S. Chen, P. T. Hammond and S. H. Yang, Carbon, 2011, 49, 457–467 CrossRef CAS PubMed.
- J. B. Goodenough, H. D. Abruńa and M. V. Buchanan, Report of the basic energy sciences workshop on electrical energy storage, US department of Energy, Washington, 2007 Search PubMed.
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845–854 CrossRef CAS PubMed.
- Z. S. Wu, D. W. Wang, W. Ren, J. Zhao, G. Zhou, F. Li and H. M. Cheng, Adv. Funct. Mater., 2010, 20, 3595–3602 CrossRef CAS.
- L. Lai, H. Yang, L. Wang, B. K. The, J. Zhong, H. Chou, L. Chen, W. Chen, Z. Shen, R. S. Ruoff and J. Lin, ACS Nano, 2012, 6, 5941–5951 CrossRef CAS PubMed.
- M. Inagaki, H. Konno and O. Tanaike, J. Power Sources, 2010, 195, 7880–7903 CrossRef CAS PubMed.
- A. G. Pandolfo and A. F. Hollenkamp, J. Power Sources, 2006, 157, 11–27 CrossRef CAS PubMed.
- C. C. Hu and T. W. Tsou, Electrochem. Commun., 2002, 4, 105–109 CrossRef CAS.
- L. L. Zhang, S. Zhao, X. N. Tian and X. S. Zhao, Langmuir, 2010, 26, 17624–17628 CrossRef CAS PubMed.
- A. K. Saker and J. D. Hong, Langmuir, 2012, 28, 12637–12646 CrossRef PubMed.
- J. P. Ferraris, M. M. Eissa, I. D. Brotherston and D. C. Loveday, Chem. Mater., 1998, 10, 3528–3535 CrossRef CAS.
- A. Pendashteh, M. F. Mousavi and M. S. Rahmanifar, Electrochim. Acta, 2013, 88, 347–357 CrossRef CAS PubMed.
- Y. Chen, X. Zhang, D. Zhang, P. Yu and Y. Ma, Carbon, 2011, 49, 573–580 CrossRef CAS PubMed.
- M. J. Allen, V. C. Tung and R. B. Kaner, Chem. Rev., 2010, 110, 132–145 CrossRef CAS PubMed.
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228–240 RSC.
- S. Pei and H. M. Cheng, Carbon, 2010, 50, 3210–3228 CrossRef PubMed.
- S. Stankovich, D. A. Dikin, G. H. B. Demmett, K. M. Kohlhaas, E. J. Zimney, E. A. Stach, R. D. Piner, S. T. Nguyen and R. S. Ruoff, Nature, 2006, 442, 282–286 CrossRef CAS PubMed.
- W. H. Khoh and J. D. Hong, Colloids Surf., A, 2013, 436, 104–112 CrossRef CAS PubMed.
- Y. Zhu, S. Murali, W. Cai, X. Li, J. W. Suk, J. R. Potts and R. S. Ruoff, Adv. Mater., 2010, 22, 3906–3924 CrossRef CAS PubMed.
- Z. F. Li, H. Zhang, Q. Liu, L. Sun, L. Stanciu and J. Xie, ACS Appl. Mater. Interfaces, 2013, 5, 2685–2691 CAS.
- Q. WU, Y. Xu, Z. Yao, A. Liu and G. Shi, ACS Nano, 2010, 4, 1963–1970 CrossRef CAS PubMed.
- J. Yan, T. Wei, B. Shao, F. Ma, Z. Fan, M. Zhang, C. Zheng, Y. Shang, W. Qian and F. Wei, Carbon, 2010, 48, 1731–1737 CrossRef CAS PubMed.
- C. Xiang, M. Li, M. Zhi, A. Manivannan and N. Wu, J. Mater. Chem., 2012, 22, 19161–19167 RSC.
- M. S. A. S. Shah, A. R. Park, K. Zhang, J. H. Park and P. J. Yoo, ACS Appl. Mater. Interfaces, 2012, 4, 3893–3901 Search PubMed.
- D. Wang, X. Li, J. Chen and X. Tao, Chem. Eng. J., 2012, 198–199, 547–554 CrossRef CAS PubMed.
- A. Ramadoss and S. J. Kim, Carbon, 2013, 63, 434–445 CrossRef CAS PubMed.
- M. C. Yang, Y. Y. Lee, B. Xu, K. Powers and Y. S. Meng, J. Power Sources, 2012, 207, 166–172 CrossRef CAS PubMed.
- A. K. Mishra and S. Ramaprabhu, J. Phys. Chem. C, 2011, 115, 14006–14013 CAS.
- J. Liu, J. Sun and L. Gao, J. Phys. Chem. C, 2010, 114, 19614–19620 CAS.
- J. Liu, J. Sun and L. Gao, Nanoscale, 2011, 3, 3616–3619 RSC.
- J. Liu, L. Zhang, H. B. Wu, J. Lin, Z. Shen and X. W. Lou, Energy Environ. Sci., 2014, 7, 3709–3719 CAS.
- Y. Xu, Z. Lin, X. Huang, Y. Liu, Y. Huang and X. Duan, ACS Nano, 2013, 7, 4042–4049 CrossRef CAS PubMed.
- M. Dahl, Y. Liu and Y. Yin, Chem. Rev., 2014, 114, 9853–9889 CrossRef CAS PubMed.
- L. Tian, P. Anilkumar, L. Cao, C. Y. Kong, M. J. Meziani, H. Qian, L. M. Veca, T. J. Thorne, K. N. Tackett, T. Edwards and Y. P. Sun, ACS Nano, 2011, 5, 3052–3058 CrossRef CAS PubMed.
- C. Xu, A. Cui, Y. Xu and X. Fu, Carbon, 2013, 62, 465–471 CrossRef CAS PubMed.
- C. Xu, A. Cui, Y. Yuan, Z. Chen, R. Yuan and X. Fu, J. Mater. Sci., 2013, 48, 3428–3435 CrossRef CAS.
- Y. Li, J. Wang, X. Li, D. Geng, R. Li and X. Sun, Chem. Commun., 2011, 47, 9438–9440 RSC.
- Y. Si and E. T. Samulski, Nano Lett., 2008, 8, 1679–1682 CrossRef CAS PubMed.
- M. Acik, G. Lee, C. mattevi, M. chhowalla, K. Cho and Y. J. Chabal, Nat. Mater., 2010, 9, 840–845 CrossRef CAS PubMed.
- H. Liu, L. Zhang, Y. Guo, C. Cheng, L. Yang, L. Jiang, G. Yu, W. Hu, Y. Liu and D. Zhu, J. Mater. Chem. C, 2013, 18, 3104–3109 RSC.
- G. K. Ramesha and S. Sampath, J. Phys. Chem. C, 2009, 113, 7985–7989 CAS.
- A. C. Ferrari and J. Robertson, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 14095–14107 CrossRef CAS.
- F. Dong, S. Guo, H. Wang, X. Li and Z. Wu, J. Phys. Chem. C, 2011, 115, 13285–13292 CAS.
- B. H. Wee and J. D. Hong, Adv. Funct. Mater., 2013, 23, 4657–4666 CrossRef CAS.
- Y. Liang, H. Wang, H. S. Casalongue, Z. Chen and H. Dai, Nano Res., 2010, 3, 701–705 CrossRef CAS PubMed.
- B. Y. Xia, H. B. Wu, J. S. Chen, Z. Wang, X. Wang and X. W. Lou, Phys. Chem. Chem. Phys., 2012, 14, 473–476 RSC.
- W. W. Liu, X. B. Yan, J. W. Lang, C. Peng and Q. J. Xue, J. Mater. Chem., 2012, 22, 17245–17253 RSC.
- Y. Wang, Z. Shi, Y. Huang, Y. Ma, C. Wang, M. Chen and Y. Chen, J. Phys. Chem., 2009, 113, 13103–13107 CAS.
- V. Khomenko, E. R. Pinero, E. Frackowiak and F. Beguin, Appl. Phys. A, 2006, 82, 567–573 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The CV curve and galvanostatic charge/discharge curve of rGT electrode reduced at different temperature. See DOI: 10.1039/c4ra12980f |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.