
Polymer–paclitaxel conjugates based on disulfide linkers for controlled drug release
Wulian Chena,
Luqman Ali Shahb,
Li Yuana,
Mohammad Siddiqb,
Jianhua Hua and
Dong Yang*a
aState Key Laboratory of Molecular Engineering of Polymers, Department of Macromolecular Science, Fudan University, Shanghai 200433, China. E-mail: yangdong@fudan.edu.cn; Fax: +86-21-65640293; Tel: +86-21-65643575
bDepartment of Chemistry, Quaid-i-Azam University, Islamabad 45320, Pakistan
First published on 22nd December 2014
Abstract
A novel redox-responsive polymer–drug conjugate (PDC) based on hydrophilic diblock copolymer covalently linked paclitaxel (PTX) via a disulfide linker was prepared and evaluated for intracellular drug delivery. The well-defined hydrophilic diblock copolymer, PEG-b-PHEMA, was synthesized via atom transfer radical polymerization of 2-(trimethylsilyloxyl)ethyl methacrylate (HEMA-TMS), using PEG-Br as a macroinitiator and CuBr/PMDETA as the catalytic system, followed by selectively hydrolyzing the trimethylsilane group to hydroxyl groups. Utilizing the hydroxyl groups as an active reaction site, paclitaxel was covalently conjugated onto the backbone of the diblock copolymer, with a disulfide linker as a spacer to bridge the copolymer and PTX, and the loading content of paclitaxel was 18.4 wt%. Due to the different solubility of segments in the polymer–drug conjugate, the amphiphilic PEG-b-P(HEMA-PTX) could self-assemble into spherical micelles in aqueous solution, with hydrophobic PTX as core and hydrophilic PEG chains as shell. The in vitro cytotoxicity experimental results showed that the diblock copolymer was biocompatible, with no obvious cytotoxicity, whereas the PEG-b-P(HEMA-PTX) conjugate showed glutathione-dependent cytotoxicity with higher cellular proliferation inhibition against glutathione monoester pretreated HeLa cells than that of the nonpretreated HeLa cells. We are convinced that polymer–drug conjugates based on disulfide linkers will be a promising platform for targeted intracellular controlled drug delivery in cancer therapy.
1. Introduction
Amphiphilic block copolymers can self-assemble into micelles with core–shell structure in aqueous solution, and have shown great potential in applications as nanocarriers of chemotherapeutic drugs and imaging agents.1–5 Up to date, most of the polymeric micelles have been loaded with drugs via non-covalent interactions.6 Although these controlled drug delivery systems (CDDSs) have increased water solubility, reduced toxicity, prolonged circulation half-life and improved accumulation in tumor tissues due to the enhanced permeability and retention (EPR) effect, these CDDSs have a drug burst release effect, which is the main reason for toxicity and side effects and dramatically decreases the therapeutic efficacy.7–10 Thus, polymer–drug conjugates (PDCs) based on polymers with covalently loaded of drug, have been proposed to overcome the drug burst release. As a special class of functional polymeric materials, PDCs have labile linkages between drug molecules and polymers, and therefore can allow drug molecules to be released through the cleavage of linkages under a specific stimulus like pH, reducing agents and light.11–15 As compared with polymeric micelle CDDSs loading drug via physical entrapment, PDCs typically have well-controlled drug loadings and continuous release without burst release effect.16–19 Noting that some anticancer drugs like paclitaxel (PTX) are highly hydrophobic, PDCs micelles can be easily constructed by using water-soluble polymer to covalently load hydrophobic drug molecules.As well-known, among the vast of hydrophilic and biocompatible polymers, poly(ethylene glycol) (PEG)20 and poly(2-hydroxyethyl methacrylate) (PHEMA)21 are widely applied in biochemical and biomedical fields. Although PEG could effectively protect the core against the external biological media, inhibit non-specific protein absorption, and increase the plasma clearance half-life, it lacked reactive groups covalently linked functional molecules. Relative to PEG, PHEMA contains reactive hydroxyls covalently linked functional molecules to its surfaces, thus, endowing polymer with controlled drug release and targeted functions. The copolymers containing PEG and PHEMA segments have been synthesized in the past decade. Matyjaszewski et al. first synthesized linear PHEMA by atom transfer radical polymerization (ATRP), following esterification reactions between pentynoic acid and the hydroxyl groups. Utilizing the alkynyl groups, molecular brushes, PHEMA-g-PEG, were obtained by click reaction.22 Xu et al. prepared well-defined copolymer, PHEMA-b-P(DMAEMA)-b-PEG-b-P(DMAEMA)-b-PHEMA, via ATRP of 2-(dimethylamino)ethyl methacrylate (DMAEMA) and HEMA, using Br-PEG-Br as macroinitiator, which was used as gene carrier in gene delivery systems.23 Chen et al. prepared amphiphilic toothbrush like copolymer, PEG-b-P(HEMA-g-PCL), via the ring-opening polymerization of ε-caprolactone (CL) initiated by PEG-b-PHEMA macromolecular initiators.24 However, the work that drugs are conjugated to hydrophilic PEG-b-PHEMA copolymer via the hydroxyl groups of PHEMA segment is rarely reported.
Commonly, the drug and the polymer are conjugated via relatively stable linkers (e.g. amide and ester bonds).25 However, it should be noted that overly stable linkers are not ideal either because the release of drugs may be prohibited after PDCs arrive at the target site, resulting in low drug efficacy.26,27 In addition, the drug released from PDCs should keep the original structure to achieve the drug therapy and avoid the undesirable side effect. In recent times, the disulfide bond, an extremely valuable functional group in a variety of chemical and biological agents, is being widely investigated in the past decade.28–33 In the human body, intracellular GSH concentration is in the millimolar range (approximately 1–10 mM). That is 2–3 orders higher than its concentration in common fluids outside cells (usually micromolar, ∼10 μM), such as plasma and other body fluids.34,35 Moreover, it has been found that intracellular GSH concentration is several times higher than that in the corresponding normal cells.36–38 Based on such diversity, the disulfide bonds are stable under physiological conditions in the circulation, but can quickly be cleaved in a highly reductive environment within cells, achieving controlled intracellular rapid release. Therefore PDCs with a disulfide linker as spacer to bridge polymer and drug is an artful approach to address the extracellular stability and the intracellular drug release dilemma.
Recently, Ojima et al. have reported a novel disulfide linker which could react with reducing agents to release drug without changing the chemical structure of drug.39 Herein, we utilized this disulfide linker to construct a novel CDDS based on hydrophilic diblock copolymer covalently linked to PTX via a disulfide bond. The well-defined hydrophilic diblock copolymer, PEG-b-PHEMA, was synthesized via the atom transfer radical polymerization (ATRP) of 2-(trimethylsilyloxyl)ethyl methacrylate (HEMA-TMS), using PEG-Br as macroinitiator and CuBr/PMDETA as a catalytic system, followed by selectively hydrolyzing trimethylsilane group to hydroxyl groups. Utilizing the hydroxyl groups as an active reaction site, paclitaxel could be covalently linked to the backbone of copolymer via disulfide bond (Scheme 1), and the loading content of paclitaxel could reach up to 18.4 wt%. The chemical structures of the diblock copolymer and PTX–polymer conjugate were characterized by 1H NMR, GPC, FT-IR, and so on. The in vitro cellular cytotoxicity experiments were performed to evaluate the biocompatibility of the copolymer, and the intracellular drug release was also evaluated against a HeLa human cervical carcinoma cell line, which were pretreated with different concentrations of glutathione monoester (GSH-OEt) and had different concentrations of glutathione in the cytoplasm.
 | ||
| Scheme 1 Schematic illustration of the synthesis of polymer–drug conjugates of PEG-b-P(HEMA-PTX) via an disulfide linker and its drug release mechanism. | ||
2. Experimental section
2.1 Materials
PTX was purchased from Beijing Huafeng United Technology Co., Ltd. 2-(Trimethylsilyloxy)-ethyl methacrylate (HEMA-TMS, 96%, Aldrich) was passed through a neutral alumina column to remove inhibitor. Triethylamine (TEA, 99.5%, Aldrich) was dried over CaH2 and distilled at 110 °C. Toluene (99%, Aldrich) was refluxed with sodium beads/benzophenone complex and distilled until the solution turned purple. N,N′-Dimethylformamide (DMF) was dried over CaH2 and distilled under reduced pressure. N,N,N′,N′′,N′′′-Pentamethyldiethyldiethylenetriamine (PMDETA, 98%, Aldrich), copper(I) bromide (CuBr, 99.999%, Aldrich), 2-bromoisobutyl bromide (BiBB, 98%, Acros), glutathione monoethyl ester (GSH-OEt, Aldrich), 1-ethyl-3-(3-dimethyllaminopropyl) and N,N-diisopropylethylamine (DIPEA, 99%, Aldrich) were used as received without further purification.2.2 Preparation of PEG-b-PHEMA
PEG-Br macroinitiator was prepared according to previous literatures.40–43 GPC: Mn = 6000 g mol−1, polydispersity index (PDI) = 1.02. 1H NMR (CDCl3): δ (ppm): 1.84 (6H, C(CH3)2Br), 3.37 (3H, OCH3), 3.63 (4H, OCH2CH2). PEG-b-P(HEMA-TMS) diblock copolymer was obtained by ATRP of HEMA-TMS using PEG-Br as the macroinitiator and CuBr/PMDETA as a catalytic system. Typically, a dry 25 mL Schlenk flask (flame-dried under vacuum prior to use) was charged with PEG-Br (1.0 g, 0.2 mmol Br group) and CuBr (28.7 mg, 0.2 mmol). After three evacuate-nitrogen-filling cycles, HEMA-TMS (2.2 mL, 10 mmol), dry toluene (10 mL), and PMDETA (50 μL, 0.24 mmol) were introduced via a gastight syringe. The mixture was degassed with three freeze–evacuate–thaw cycles, followed by immersing the flask into an oil bath preset at 80 °C. The polymerization lasted for 12 h and was terminated by immersing the flask into liquid N2. Then, the mixture was diluted with THF and passed through a short neutral alumina column to remove the residual copper catalyst. The filtrate was concentrated under vacuum and precipitated into cold petroleum ether twice. The white product, 2.3 g, PEG-b-P(HEMA-TMS), was obtained after drying in vacuo at 40 °C overnight (yield of 83.3%). GPC: Mn = 19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300 g mol−1, PDI = 1.21. 1H NMR (CDCl3): δ (ppm): 0.13 (9H, Si(CH3)3), 0.85–1.05 (3H, CH3CCH2 of P(HEMA-TMS)), 1.75–1.98 (2H, CH2CCH3 of P(HEMA-TMS)), 3.63 (4H, OCH2CH2 of PEG), 3.75 (2H, CH2OSi(CH3)3 of P(HEMA-TMS)), 3.98 (2H, COOCH2 of P(HEMA-TMS)).
300 g mol−1, PDI = 1.21. 1H NMR (CDCl3): δ (ppm): 0.13 (9H, Si(CH3)3), 0.85–1.05 (3H, CH3CCH2 of P(HEMA-TMS)), 1.75–1.98 (2H, CH2CCH3 of P(HEMA-TMS)), 3.63 (4H, OCH2CH2 of PEG), 3.75 (2H, CH2OSi(CH3)3 of P(HEMA-TMS)), 3.98 (2H, COOCH2 of P(HEMA-TMS)).
PEG-b-P(HEMA-TMS) (0.6 g) was dissolved into THF/methanol (30 mL/10 mL), and then 10 drops of hydrochloric acid (1 mol L−1) were added. The mixture was stirred at room temperature for 48 h, followed by concentrating and precipitating in cold hexane. After repeated purification by dissolving in THF and precipitating in cold hexane, 0.4 g of the white powder, PEG-b-PHEMA, was obtained after drying in vacuo at 40 °C overnight (yield of 82.6%). 1H NMR (CDCl3): δ (ppm): 0.90–1.09 (3H, CH3CCH2 of PHEMA), 1.93–2.05 (2H, CH2CCH3 of PHEMA), 3.60 (4H, OCH2CH2 of PEG), 3.75 (2H, CH2OH of PHEMA), 4.04 (2H, COOCH2 of PHEMA), 5.03 (1H, CH2CH2OH of PHEMA).
2.3 Preparation of PEG-b-(PHEMA-PTX)
PTX was covalently conjugated onto PEG-b-PHEMA via an esterification reaction using DIPEA as a catalyst at room temperature. Typically, PEG-b-PHEMA (50 mg, 0.23 mmol of hydroxyl group), PTX-linker-OSu (70 mg, 58.1 μmol) prepared according to ref. 30 and 39, DIPEA (50 μL) and anhydrous DMF (5 mL) were added into a 10 mL of flask. The mixture was stirred at room temperature for 24 h. After repeated purification by dissolving in DMF and precipitating in ether, the crude product was isolated by enough dialysis against deionized water using a dialysis bag with a cut-off molecular weight of 3.5 kDa. Finally, a gray powder (61.3 mg, yield of 51.1%), PEG-b-P(HEMA-PTX) was obtained by freeze-drying.For preparation of polymer–drug conjugate micelles, 2 mg PEG-b-P(HEMA-PTX) was dissolved into 0.5 mL DMSO, and then 2.5 mL of deionized water was added dropwise under stirring. After further stirring for 2 h, DMSO was removed by enough dialysis against deionized water using a dialysis bag with a cut-off molecular weight of 3.5 kDa to afford a micellar solution.
2.4 In vitro drug release
The PTX release from polymer–drug conjugate was investigated at 37 °C in two different medias, (a) PBS of pH = 7.4 with 10 mM of cysteamine and PBS of pH = 7.4 without cysteamine. Typically, 5 mg of PEG-b-P(HEMA-PTX) was dissolved in 10 mL of deionized water. Then, 5 mL of solution was transferred to a dialysis bag with a cut-off molecular weight of 3.5 kDa and immersed in 100 mL of two different medias with constant shake at 37 °C. At desired time intervals, 10 mL of solution was taken out for UV measurement and replenished with an equal volume of corresponding fresh media.2.5 In vitro cell assay
The cytotoxicity assay of PEG-b-PHEMA against HeLa cervical carcinoma cells was evaluated by a CCK-8 kit assay. The cells were seeded in 96-well plates with a density of 1 × 104 cell per well and incubated at 37 °C in an atmosphere containing 5% CO2 for 24 h to allow cell attachment. Then, the medium was replaced with a fresh medium containing the indicated concentration of PEG-b-PHEMA. After incubation for 48 h, the medium was aspirated and replaced by 100 μL of fresh medium containing 10 μL of CCK-8. The cells were incubation for another 1 h at 37 °C in dark. Afterward, the absorbance at a wavelength of 450 nm of each well was measured using a microplate reader.The intracellular drug release of PEG-b-P(HEMA-PTX) at different concentrations of glutathione in the cytoplasm was studied. The cells were treated with medium solution of GSH-OEt for another 2 h, and then the medium was aspirated and replaced by fresh medium containing PEG-b-P(HEMA-PTX). Cells without pre-treatment were used as the control. After 48 h of incubation time, the cells were then washed two times with PBS. The following procedure of the cell viability determined by CCK-8 assays was same as described above.
2.6 Characterizations
1H NMR spectra were recorded on a Bruker Avance 500 spectrometer using CDCl3 and DMSO-d6 as solvents, trimethylsilane was used as an internal standard. All FT-IR spectra were recorded as KBr pellets on a Magna 550 FT-IR spectrophotometer with a resolution of 4 cm−1. The molecular weight and molecular weight distribution were determined by gel permeation chromatography (GPC). The GPC was performed in THF at 35 °C with an elution rate of 1.0 mL min−1 on Aginent 1100 equipped with a G1310A pump, a G1362A refractive index detector, and a G1314A variable wavelength detector. Polystyrene standard samples were employed for the GPC calibration. Transmission electron microscopy (TEM) images were obtained on a JEOL JEM 2100 F transmission electron microscope, and samples for TEM measurements were made by casting one drop of the sample's solution on a carbon copper grid. The size distribution of the micelles was measured by dynamic light scattering (DLS) using a Malvern autosize 4700 instrument. Elemental analysis was carried out on a Carlo-Erba 1106 system.3. Results and discussion
HEMA is a very polar monomer and the propagating radicals of HEMA are stable and propagate slowly, which dramatically affect the ATRP of HEMA and function of the catalytic species. Matyjaszewski successfully synthesized PHEMA with controllable molecular weight and molecular weight distribution via ATRP using HEMA-TMS as monomer.44 In this paper, PEG-b-P(HEMA-TMS) diblock copolymer was firstly obtained via ATRP of HEMA-TMS using PEG113-Br as macroinitiator and CuBr/PMDETA as catalytic system in toluene at 80 °C. After hydrolyzing the protected group of TMS, the required diblock copolymer, PEG-b-PHEMA was obtained. A typical GPC trace of PEG-b-P(HEMA-TMS) was shown in Fig. 1, it appeared a monomodal elution peak and a clear shift to the higher molecular weight side, compared with that of PEG-Br macroinitiator. The molecular weight distributions (Mw/Mn = 1.21) was also narrow according to the results of GPC. These observations clearly suggested that ATRP of HEMA-TMS was really controlled well. The chemical structure of PEG-b-P(HEMA-TMS) diblock copolymer was characterized by 1H NMR. As shown in the 1H NMR spectra of Fig. 2A, characteristic signals of PEG and P(HEMA-TMS) were all present. The DP of P(HEMA-TMS) was determined to be 49 as indicated in 1H NMR analysis from the signal integration ratio of peaks d at 0.13 ppm (methyl silane protons of P(HEMA-TMS)) relative to peak a at 3.60 ppm (CH2CH2O of PEG). The experimental DP was consistent with the theoretical DP (DPt = 50). Trimethylsilane group was widely used for the protection of hydroxyl group, and could be readily hydrolyzed under acidic condition. The hydrolyzed product was characterized by 1H NMR to confirm the selective hydrolysis of trimethylsilane group without destroying the backbone. As seen from Fig. 2B, the peak of nine protons of trimethylsilane group at 0.13 ppm was completely disappeared after hydrolysis, and a new peak, assigned to the proton of hydroxyl group, appeared at 5.03 ppm. The result indicated that the PEG-b-P(HEMA-TMS) was selectively hydrolyzed to PEG-b-PHEMA. | ||
| Fig. 1 GPC traces of (A) PEG-Br and (B) PEG-b-P(HEMA-TMS) with THF as the elute. | ||
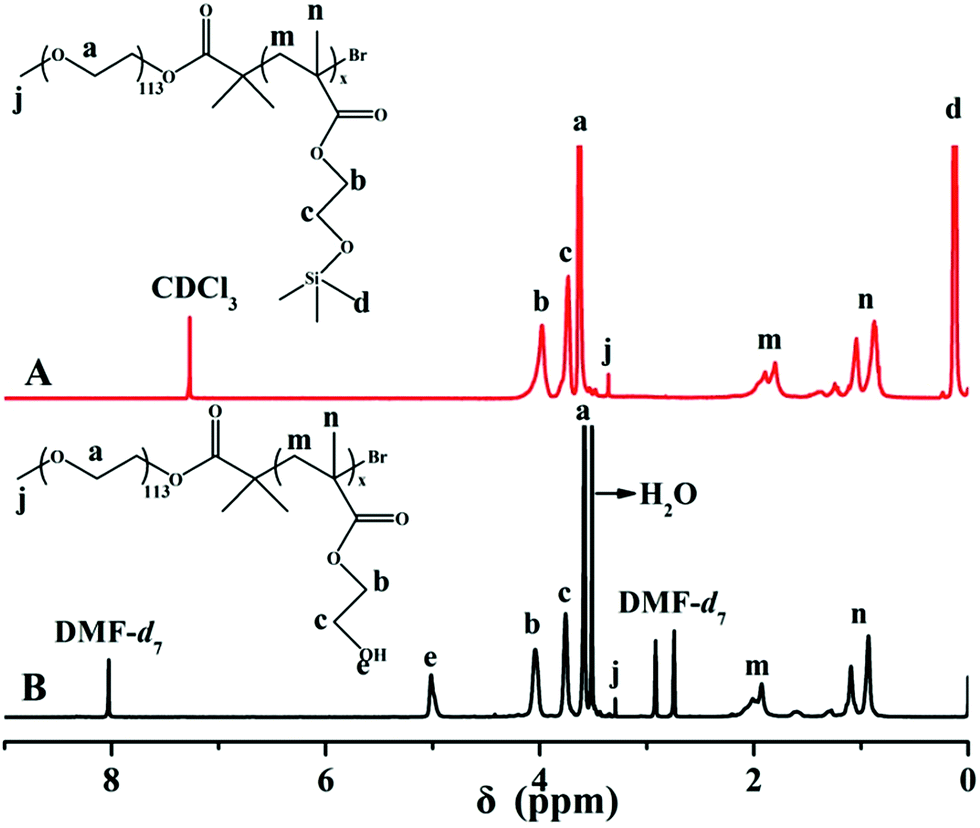 | ||
| Fig. 2 1H NMR spectra of (A) PEG-b-P(HEMA-TMS) in CDCl3 and (B) PEG-b-PHEMA in DMF-d7. | ||
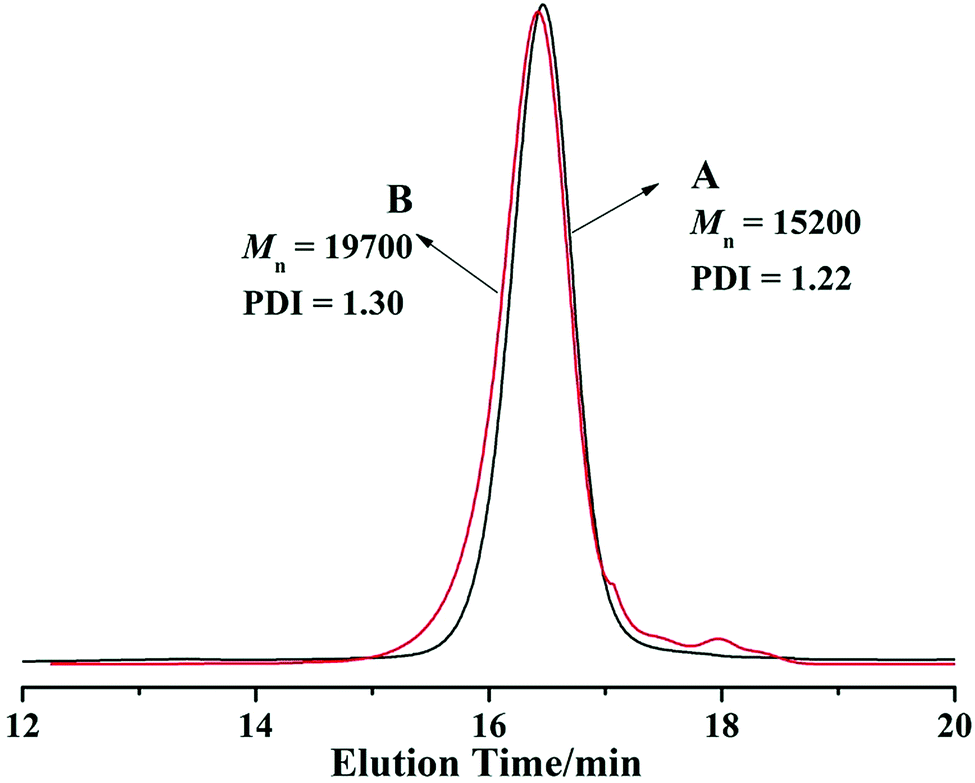 | ||
| Fig. 3 GPC traces of (A) PEG-b-PHEMA and (B) PEG-b-P(HEMA-PTX) with DMF as the elute. | ||
The PTX-linker-OSu containing disulfide bond with redox was synthesized according to previous literatures30,39 and could easily react with amino or hydroxyl groups. Thus, PTX was covalently loaded onto PEG-b-PHEMA via a disulfide bond. Due to strong interaction between PHEMA segment and GPC column with THF as elute, the GPC traces of PEG-b-PHEMA and PEG-b-P(HEMA-PTX) were obtained with DMF as elute. As seen from in Fig. 3, after PTX-linker loaded onto PEG-b-PHEMA, PEG-b-P(HEMA-PTX) showed a higher molecular weight than PEG-b-PHEMA. The chemical structure of resulting polymer–drug conjugate PEG-b-P(HEMA-PTX) was characterized by 1H NMR. Fig. 4 shows representative 1H NMR spectra of PEG-b-PHEMA, PEG-b-P(HEMA-PTX) and PTX-linker-OSu (hydroxysuccinimide), respectively. It is found that the characteristic signals of diblock polymer and prodrug molecule were all present in 1H NMR spectrum of PEG-b-P(HEMA-PTX). The result confirmed that the prodrug molecule PTX-linker-OSu was successfully covalently loaded onto PEG-b-PHEMA. Meanwhile, the above three samples were also characterized by FT-IR and the results were shown in Fig. 5. Although most characteristic peaks of PEG-b-PHEMA were the same with that of PTX-linker-OSu, it is found that two new peaks at 1524 and 713 cm−1, which were assigned to characteristic peaks of PTX-linker-OSu, appeared in FT-IR spectrum of PEG-b-P(HEMA-PTX) but did not appeared in FT-IR spectrum of PEG-b-PHEMA in Fig. 5. All of these results suggested the successful preparation of polymer–drug conjugate PEG-b-P(HEMA-PTX).
 | ||
| Fig. 4 1H NMR spectra of (A) PEG-b-PHEMA in DMF-d7, (B) PEG-b-P(HEMA-PTX) in DMSO-d6 and (C) PTX-linker-OSu in CDCl3. | ||
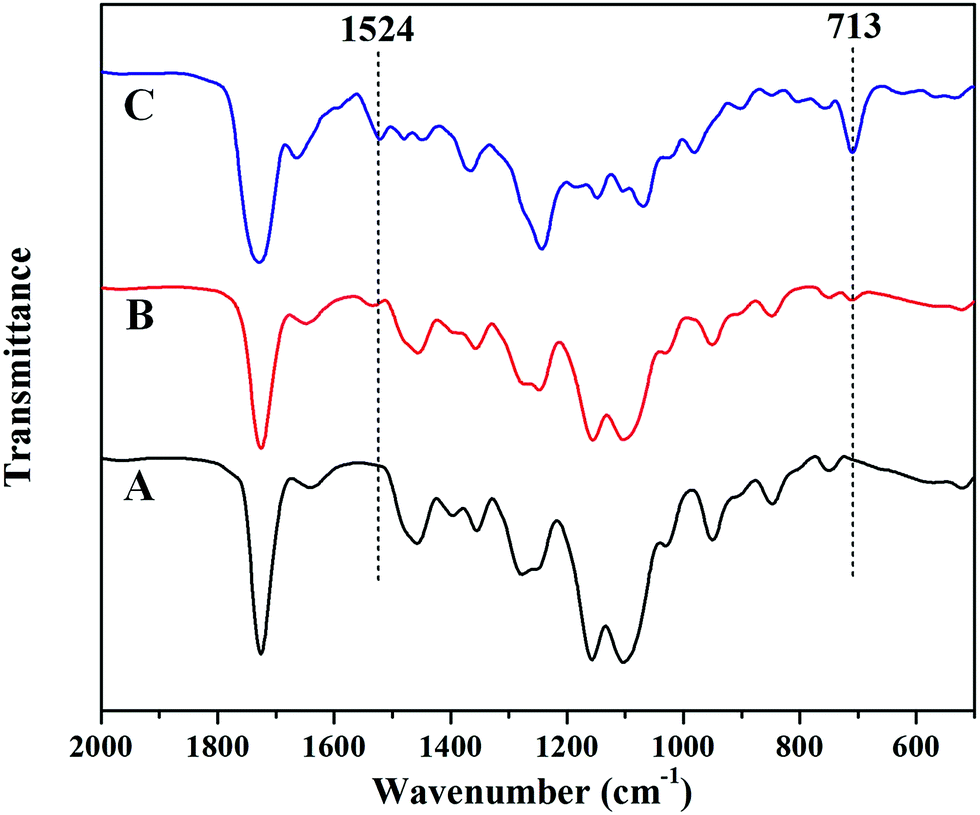 | ||
| Fig. 5 Enlarged FT-IR spectra of (A) PEG-b-PHEMA, (B) PEG-b-P(HEMA-PTX) and (C) PTX-linker-OSu. | ||
After hydrophobic PTX covalently loaded on water soluble diblock copolymer PEG-b-P(HEMA), the resulting polymer–drug conjugate PEG-b-P(HEMA-PTX) was amphiphilic, and could self-assemble into micelles with hydrophobic PTX as core and hydrophilic PEG chain as shell in an aqueous solution. As seen from the TEM image of Fig. 6A, the micelles were spherical and with an average diameter of approximately 200 nm. The results of DLS in Fig. 6B showed that PEG-b-P(HEMA-PTX) micelles exhibited an intensity-average hydrodynamic diameter of 304 nm. And the polydispersity index (PDI) of the micelles is 0.106. Compared with the result of DLS, the average diameter of PEG-b-P(HEMA-PTX) micelles determined by TEM were smaller. This is because the TEM technique determines nanoparticles dimension in the dry state, whereas DLS reports intensity-average dimension in solution. Moreover, the outer shell of PEG are invisible under TEM observation. The illustration of water-soluble PEG-b-P(HEMA) and amphiphilic PEG-b-P(HEMA-PTX) in water was also shown in Fig. 6B. It is obviously observed that the solution of PEG-b-P(HEMA) was transparent, but the solution of PEG-b-P(HEMA-PTX) was emulsion, which indicated that PEG-b-P(HEMA-PTX) could self-assemble into micelles in water.
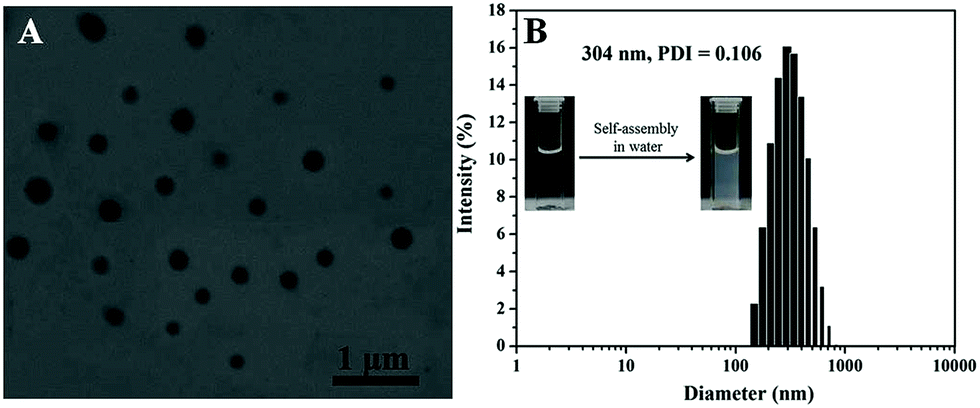 | ||
| Fig. 6 (A) TEM image of PEG-b-P(HEMA-PTX) micelles self-assembled in water. (B) Hydrodynamic diameter distribution of PEG-b-P(HEMA-PTX) micelles. The inset is the digital photograph of PEG-b-PHEMA solution and PEG-b-P(HEMA-PTX) micelles in water. | ||
To ascertain the core–shell structure, 1H NMR characterizations of PEG-b-P(HEMA-PTX) using D2O and DMSO-d6 as solvents were performed. As can be seen from Fig. 7, the 1H NMR spectrum of PEG-b-P(HEMA-PTX) using DMSO-d6 as a solvent (Fig. 7B) showed distinguished characteristic PTX peaks at 1.0 to 2.2 ppm, and aromatic signals at 7 to 8 ppm. While using D2O as a solvent (Fig. 7A), these characteristic signals disappeared, indicating that the hydrophobic PTX was in the inner core of the micelles. This is because that the amphiphilic polymer–drug conjugate chain could well extend in DMSO-d6, but form a spherical micelle with PTX as core and PEG chains as shell in D2O. The loading content of PTX on the diblock copolymer was determined by the element analysis. The element analysis results showed that the sulfur content in PEG-b-P(HEMA-PTX) conjugate was 1.38%, thus, the loading content of PTX on the diblock copolymer was about 18.4%.
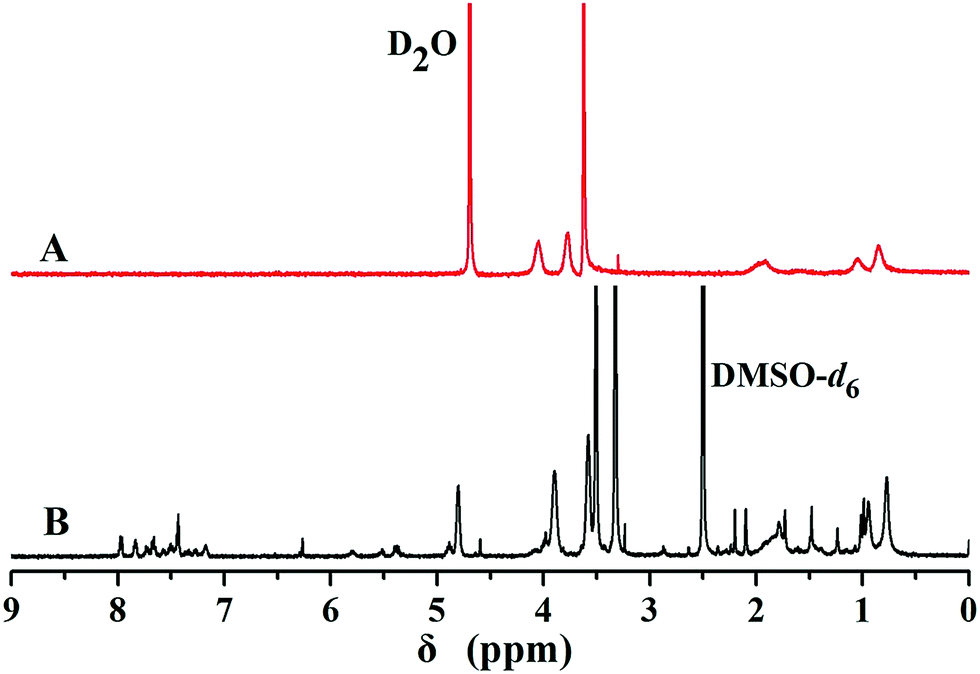 | ||
| Fig. 7 1H NMR spectra of PEG-b-P(HEMA-PTX) in (A) D2O and (B) DMSO-d6. | ||
The disulfide bonds can be cleaved through the thiol–disulfide exchange reaction under reduced environment. To confirm the reduction sensitivity of PEG-b-P(HEMA-PTX), the in vitro drug release profiles were investigated in PBS with 10 mM of cysteamine and without addition of cysteamine, respectively. As seen from Fig. 8, the amount of PTX released from PEG-b-P(HEMA-PTX) was 21.3% in PBS without cysteamine within 60 h. However, 72.3% of PTX was release from PEG-b-P(HEMA-PTX) in PBS with 10 mM of cysteamine within 60 h. These results indicated that PEG-b-P(HEMA-PTX) polymer–drug conjugate was sufficiently stable under physiological condition, while it could rapidly release PTX under reduced condition, such as intracellular GSH in cancer cell. Therefore, redox-responsive polymer–drug conjugate have obvious advantage in CDDSs.
 | ||
| Fig. 8 In vitro PTX release profile recorded for PEG-b-P(HEMA-PTX) conjugate at (■) PBS without cysteamine and (●) PBS with 10 mM of cysteamine. | ||
It is very important to evaluate the potential toxicity of polymeric materials for drug delivery application. The in vitro cytotoxicity of PEG-b-P(HEMA) against HeLa (human tumor cells) and HEK-293 (human normal cells) with different concentrations was studied by the CCK-8 assays. As seen from Fig. 9, PEG-b-P(HEMA) showed no obvious cytotoxic effect on HeLa cells at a concentration ranged from 0.01 to 5.0 mg mL−1. Furthermore, HEK-293 cell could also tolerate the treatment with PEG-b-P(HEMA) at 5.0 mg mL−1, and the cell viability was still above 80% after incubation for 48 h (Fig. 9). These results suggested the cytotoxicity of PEG-b-P(HEMA) was pretty low, and suitable to use as drug carrier in DDSs.
 | ||
| Fig. 9 Cell viability of HeLa cells and HEK-293 cells incubated with different concentrations of PEG-b-PHEMA for 48 h. | ||
It is interesting for us to study the intracellular reducing responsiveness and cytotoxicity of PEG-b-P(HEMA-PTX) polymer–drug conjugate in living cell. The in vitro cellular cytotoxicity of PEG-b-P(HEMA-PTX) to HeLa cells was evaluated to determine whether the conjugates were redox-responsive and had effective cytotoxicity in the intracellular environment. It is documented that GSH-OEt can penetrate cellular membranes and rapidly reach a high intracellular concentration of GSH. In this case, the cells were incubated with different concentrations of GSH-OEt for 2 h to manipulate the intracellular concentration of GSH and then incubated with different concentrations of PEG-b-P(HEMA-PTX) conjugates. The cells without GSH-OEt pretreatment were used as control. The CCK-8 assay results of these experiments were shown in Fig. 10. As shown in Fig. 10, after 48 h incubation, all the HeLa cells exhibited significant growth inhibition and the cell viability declined along with the increased concentrations of PEG-b-P(HEMA-PTX) conjugates, whether the cells pretreated by GSH-OEt or the cells without GSH-OEt pretreatment, which was due to the cleavage of disulfide linkage to release the PTX molecules under the action of cellular reduced materials. However, in contrast with the control cells, the cell viability pretreated by GSH-OEt showed lower, and the inhibition efficiency increased with the increase of GSH-OEt concentration. The distinguishing cytotoxicity of PEG-b-P(HEMA-PTX) to the HeLa cells with different treatment can also be seen from the IC50 values. The IC50 values of PEG-b-P(HEMA-PTX) conjugates against the HeLa cells at 48 h were 0.49, 0.26 and 0.17 μg mL−1, respectively, corresponding to the cells without pretreatment, pretreated by 10 mM and 20 mM GSH-OEt, respectively. From the results discussed above, we can see that the PEG-b-P(HEMA-PTX) conjugates could effectively inhibit HeLa cells proliferation and show obvious redox-responsiveness, the inhibition efficiency of PEG-b-P(HEMA-PTX) increase with the increased intracellular GSH concentration. This redox-responsiveness and drug release behavior is favorable to achieve the intracellular targeted drug delivery and release, which makes PEG-b-P(HEMA-PTX) conjugates a promising platform in cancer therapy.
 | ||
| Fig. 10 Cell viability of HeLa cells incubated with the PEG-b-P(HEMA-PTX) polymer–drug conjugate at different concentrations for 48 h. The cells were pretreated with 20 and 10 mM GSH-OEt, respectively. The untreated cells were used as a control. | ||
4. Conclusions
In this paper, a novel redox-responsive polymer–drug conjugate CDDS was constructed by using water-soluble diblock copolymer PEG-b-PHEMA to covalently load PTX via a disulfide bond with PTX loading content reaching up to 18.4%. The TEM, DLS, and 1H NMR results demonstrated that the polymer–drug conjugate PEG-b-P(HEMA-PTX) could self-assemble into spherical micelles in aqueous solution, with hydrophobic PTX as core and hydrophilic PEG chains as shell. In vitro cell assays demonstrated that PEG-b-PHEMA as the drug carrier was biocompatible and suitable to use as drug carrier. The intracellular drug delivery of the PEG-b-P(HEMA-PTX) conjugate was investigated against HeLa cells, and the results suggested that the conjugate showed GSH-dependent cytotoxicity with higher cellular proliferation inhibition against GSH-OEt pretreated HeLa cells than that of the nonpretreated HeLa cells. Therefore, this highly versatile PEG-b-P(HEMA-PTX) conjugate, together with the redox-responsive drug release behaviors is expected to be utilized as drug delivery system for therapeutic applications.Acknowledgements
The authors thank the financial support from the National Natural Science Foundation of China (51103026, 51373035 and 51373040), the Shanghai Scientific and Technological Innovation Project (11JC1400600, 10431903000 and 124119a2400), the Shanghai Rising Star Program (12QB1402900), the Specialized Research Fund for the Doctoral Program of Higher Education (20110071120006). Mr Luqman Ali Shah also wish to acknowledge Higher Education commission (HEC) Pakistan for the financial support under the indigenous and IRSIP fellowship plan.Notes and references
- R. Gref, Y. Minamitake, M. T. Peracchia, V. Trubetskoy, V. Torchilin and R. Langer, Science, 1994, 263, 1600–1603 CAS.
- C. Feng, Y. J. Li, D. Yang, J. H. Hu, X. H. Zhang and X. Y. Huang, Chem. Soc. Rev., 2011, 40, 1282–1295 RSC.
- X. L. Hu, H. Li, S. Z. Luo, T. Liu, Y. Y. Jiang and S. Y. Liu, Polym. Chem., 2013, 4, 695–706 RSC.
- S. J. Zhai, J. Shang, D. Yang, S. Y. Wang, J. H. Hu, G. L. Lu and X. Y. Huang, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 811–820 CrossRef CAS.
- L. Yuan, W. L. Chen, J. Li, J. H. Hu, J. J. Yan and D. Yang, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 4579–4588 CrossRef CAS.
- P. Couvreur, Adv. Drug Delivery Rev., 2013, 65, 21–23 CrossRef CAS PubMed.
- R. Duncan, Nat. Rev. Drug Discovery, 2003, 2, 347–360 CrossRef CAS PubMed.
- Y. H. Bae and H. Q. Yin, J. Controlled Release, 2008, 131, 2–4 CrossRef CAS PubMed.
- Y. Matsumura and H. Maeda, Cancer Res., 1986, 46, 6387–6392 CAS.
- S. Samarajeewa, R. Shrestha, M. Elsabahy, A. Karwa, A. Li, R. P. Zentay, J. G. Kostelc, R. B. Dorshow and K. L. Wooly, Mol. Pharmaceutics, 2013, 10, 1092–1099 CrossRef CAS PubMed.
- Y. Bae, S. Fukushima, A. Harada and K. Kataoka, Angew. Chem., Int. Ed., 2003, 42, 4640–4643 CrossRef CAS PubMed.
- W. L. Chen, Y. L. Shi, H. Feng, M. Du, J. Z. Zhang, J. H. Hu and D. Yang, J. Phys. Chem. B, 2012, 116, 9231–9237 CrossRef CAS PubMed.
- Q. N. Lin, C. Y. Bao, Y. L. Yang, Q. N. Liang, D. S. Zhang, S. Y. Chen and L. Y. Zhu, Adv. Mater., 2013, 25, 1981–1986 CrossRef CAS PubMed.
- N. Larson and H. Ghandehari, Chem. Mater., 2012, 24, 840–853 CrossRef CAS PubMed.
- H. Wei, R. X. Zhuo and X. Z. Zhang, Prog. Polym. Sci., 2013, 38, 503–535 CrossRef CAS PubMed.
- Y. Yu, C. K. Chen, W. C. Law, J. Mok, J. Zou, P. N. Prasad and C. Cheng, Mol. Pharmaceutics, 2013, 10, 867–874 CrossRef CAS PubMed.
- Q. Yin, R. Tong, Y. Xu, K. Baek, L. W. Dobrucki, T. M. Fan and J. Cheng, Biomacromolecules, 2013, 14, 920–929 CrossRef CAS PubMed.
- B. Jung, Y. C. Jeong, J. H. Min, J. E. Kim, Y. J. Song, J. K. Park, J. H. Park and J. D. Kim, J. Mater. Chem., 2012, 22, 9385–9394 RSC.
- K. Nam, H. Y. Nam, P. H. Kim and S. W. Kim, Biomaterials, 2012, 33, 8122–8130 CrossRef CAS PubMed.
- S. Naahidi, M. Jafari, F. Edalat, K. Raymond, A. Khademhosseini and P. Chen, J. Controlled Release, 2013, 166, 182–194 CrossRef CAS PubMed.
- K. Knop, R. Hoogenboom, D. Fischer and U. S. Schubert, Angew. Chem., Int. Ed., 2010, 49, 2–23 CrossRef PubMed.
- H. Gao and K. Matyjaszewski, J. Am. Chem. Soc., 2007, 129, 6633–6639 CrossRef CAS PubMed.
- F. J. Xu, H. Li, J. Li, Z. Zhang, E. T. Kang and K. G. Neoh, Biomaterials, 2008, 29, 3023–3033 CrossRef CAS PubMed.
- W. Zhang, Y. Li, L. Liu, Q. Sun, X. Shuai, W. Zhu and Y. Chen, Biomacromolecules, 2010, 11, 1331–1338 CrossRef CAS PubMed.
- X. B. Xiong, A. Falamarzian, S. M. Garg and A. Lavasanifar, J. Controlled Release, 2011, 155, 248–261 CrossRef CAS PubMed.
- R. Cheng, F. Feng, F. H. Meng, C. Deng, F. J. Jan and Z. Y. Zhong, J. Controlled Release, 2011, 152, 2–12 CrossRef CAS PubMed.
- M. F. Ebbesen, D. H. Schaffert, M. L. Crowley, D. Qupicky and K. A. Howard, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 5091–5099 CrossRef CAS.
- A. N. Koo, K. H. Min, H. J. Lee, S. Lee, K. Kim, I. C. Kwon, S. H. Cho, S. Y. Jeong and S. C. Lee, Biomaterials, 2012, 33, 1489–1499 CrossRef CAS PubMed.
- S. Son, R. Namgung, J. Kim, K. Singha and W. J. Kim, Acc. Chem. Res., 2012, 45, 1100–1112 CrossRef CAS PubMed.
- L. Yuan, W. L. Chen, J. H. Hu, J. Z. Zhang and D. Yang, Langmuir, 2013, 29, 734–743 CrossRef CAS PubMed.
- B. Khorsand, G. Lapointe, C. Brett and J. K. Oh, Biomacromolecules, 2013, 14, 2103–2111 CrossRef CAS PubMed.
- S. Deshayes and A. M. Kasko, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 3531–3566 CrossRef CAS.
- H. W. Li, J. Z. Zhang, Q. Q. Tang, M. Du, J. H. Hu and D. Yang, Mater. Sci. Eng., C, 2013, 33, 3426–3431 CrossRef CAS PubMed.
- M. H. Lee, Z. G. Yang, C. W. Lim, Y. K. Lee, D. B. Sun, C. Kang and J. S. Kim, Chem. Rev., 2013, 113, 5071–5109 CrossRef CAS PubMed.
- L. Y. Tang, Y. C. Wang, Y. Li, J. Z. Du and J. Wang, Bioconjugate Chem., 2009, 20, 1095–1099 CrossRef CAS PubMed.
- A. Russo, W. Degraft, N. Friedman and J. B. Mitchell, Cancer Res., 1986, 46, 2845–2848 CAS.
- G. Saito, J. A. Swanson and K. D. Lee, Adv. Drug Delivery Rev., 2003, 55, 199–215 CrossRef CAS.
- Y. H. Tao, J. F. Han, C. T. Ye, T. Thomas and H. Y. Dou, J. Mater. Chem., 2012, 22, 18864–18871 RSC.
- J. Y. Chen, S. Y. Chen, X. R. Zhao, L. V. Kuznetsova, S. S. Wong and I. Ojima, J. Am. Chem. Soc., 2008, 130, 16778–16785 CrossRef CAS.
- L. Tong, S. Zhong, D. Yang, S. Chen, Y. J. Li, J. H. Hu, G. L. Lu and X. Y. Huang, Polymer, 2009, 50, 2341–2348 CrossRef CAS PubMed.
- L. N. Gu, C. Feng, D. Yang, Y. G. Li, J. H. Hu, G. L. Lu and X. Y. Huang, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3142–3153 CrossRef CAS.
- X. M. Song, Y. Q. Zhang, D. Yang, L. Yuan, J. H. Hu, G. L. Lu and X. Y. Huang, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 3328–3337 CrossRef CAS.
- S. Y. Liu, J. V. M. Weaver, M. Save and S. P. Armes, Langmuir, 2002, 18, 8350–8357 CrossRef CAS.
- K. L. Beers, S. Boo, S. G. Gaynor and K. Matyjaszewski, Macromolecules, 1999, 32, 5772–5776 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2015 |