
Roles of hydrogen bonds and π–π stacking in the optical detection of nitro-explosives with a luminescent metal–organic framework as the sensor†
Abstract
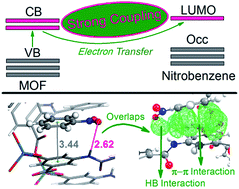
With the aids of density functional theory (DFT) and time-dependent density functional theory (TD-DFT), the explosives-detection mechanism of a typical luminescent metal–organic framework (MOF) sensor has been comprehensively studied by investigating the interactions between the framework and two analytes, namelty, benzene and nitrobenzene. By studying both the periodic crystal models and cluster models we obtained an in-depth understanding of the detecting mechanism from the viewpoint of electronic coupling between the analyte and sensor. Intermolecular electron transfer from the conduction bands of the framework to the LUMO of nitrobenzene is demonstrated to induce the luminescence quenching phenomenon observed in previous experiments. π–π stacking and hydrogen bonding interactions are found to play essential roles in this intermolecular electron transfer process. π–π stacking provides large fragment orbital overlaps between the unoccupied orbitals of the analyte and sensor, which serves as a highly efficient electron transfer bridge. Hydrogen bonds alone cannot provide enough overlaps for electron transfer but are found to reinforce the π–π stacking interactions. The cooperation of the two interactions induces facile intermolecular electron transfer which strongly quenches the luminescence of the MOF sensor. This work sheds light on the analyte–sensor interactions inside the MOF sensors and would provide valuable insights into the design of high efficient explosives-detecting MOF sensors.
- This article is part of the themed collection: Luminescence and photophysical properties of metal complexes
 Please wait while we load your content...
Please wait while we load your content...

