
Molecularly imprinted polymer nanoparticles for olanzapine recognition: application for solid phase extraction and sustained release
Abstract
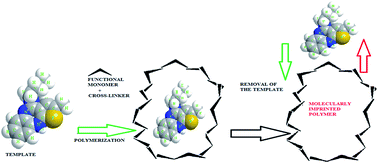
Recently, scientists have drawn more attention to polymeric nanoparticles as potential drug carriers. In this study, we synthesized high selective imprinted polymer nanoparticles using olanzapine as the template. The aim of this study was to prepare efficient imprinted polymer nanoparticles from an olanzapine template for the controlled release of olanzapine as a therapeutic drug for central nervous system (CNS) diseases at different pH values, and solid-phase extraction (SPE) as the sample clean-up technique combined with high-performance liquid chromatography (HPLC). The morphology of the nanoparticles was determined using scanning electron microscopy (SEM) images. Drug release, binding properties and dynamic light scattering (DLS) of the molecularly imprinted polymers (MIPs) were studied in this investigation. The adsorption isotherm was fitted with Langmuir and Freundlich models. The performance of the MIPs for the controlled release of olanzapine was assessed in two different media (SDS 1% and PBS). Results revealed that the MIPs have potential application in controlled drug release. Moreover, cytotoxicity of the MIP nanoparticles was measured on a NIH/3T3 cell line using a MTT method. Furthermore, the MIPs were applied to extract olanzapine from human blood plasma samples. The limit of detection (LOD) and limit of quantification (LOQ) were evaluated and were 0.18 μg L−1 and 0.39 μg L−1, respectively. These results collectively illustrate that MIP nanoparticles can be employed as an efficient technique for the extraction of the olanzapine from human plasma.
 Please wait while we load your content...
Please wait while we load your content...

