DOI:
10.1039/C4RA12690D
(Paper)
RSC Adv., 2015,
5, 13488-13494
Novel pure Pnma–P212121 ferroelastic phase transition of 1,4-diisopropyl-1,4-diazonia-bicyclo[2.2.2]octane tetra-chlorobromo-M(II) (M = Mn and Co)†
Received
19th October 2014
, Accepted 13th January 2015
First published on 13th January 2015
Abstract
Two novel phase transition compounds Dip-DABCO tetra-chlorobromo-M(II) (M = Mn and Co), (Dip-DABCO = 1,4-diisopropyl-1,4-diazonia-bicyclo[2.2.2]octane) C12H26N2·MnBrCl3 (1) and (C12H26N2)4·(CoBr1.25Cl2.75)4 (2) were synthesized, and their structures have been determined by means of single-crystal X-ray diffraction. The two compounds are isomorphous. Differential scanning calorimetry (DSC) measurements indicated that compounds 1 and 2 underwent a reversible phase transition at ca. 245.2 K with a hysteresis width of 4.4 K and at ca. 222.3 K with a hysteresis width of 5.5 K, respectively. The variable-temperature single-crystal X-ray diffraction data suggests that the phase transition was from high crystallographic symmetry with a space group of Pnma to a low-symmetry state with a space group of P212121. That is, Landau symmetry breaking occurred with a pure GPT (mmm–222). The ordering of twisting motions of the 1,4-diazoniabicylo[2,2,2]octane ring may have driven the phase transition.
Introduction
There is currently a great deal of interest in searching for new temperature-triggered solid-to-solid phase transition materials (SSPTMs) due to their potential applications in data communication, signal processing, phase shifters and environment monitoring, etc.1 Preparing new temperature-triggered molecular-based phase transition materials is crucial not only for the theoretical study of structure–property relationships but also for the exploration of novel physical properties.2,3 One of the biggest and persistent challenges in searching for phase transition materials is the rational design of novel systems.4 Constructing special types of organic–inorganic hybrid complexes may be an effective method to design ideal phase transition materials. Hybrid organic–inorganic coordination complexes possess the advantageous properties of both organic and inorganic compounds. Their topologies can be rationally designed by taking advantage of well-defined metal coordination geometries in combination with carefully chosen distorted or disordered anions, which are of benefit for the adjustment and design of ideal phase transition materials.5 Recently, hybrid inorganic–organic phase transition materials have been widely studied.6 Among them, phase transition materials containing 1,4-diazabicyclo-[2.2.2]octane (dabco) have demonstrated specific responses to external electric or temperature stimuli, and they have expanded potential areas of research of phase transition materials into the effect of the ligand. More recently, a series of metal–organic compounds assembled by 1,4-diazabicyclo-[2.2.2]octane that undergo structural transitions have been synthesized by Xiong et al.7 All these findings reveal that the dabco component may afford the potential to design and construct new functional electrically ordered materials because of its striking features, such as it being a donor–acceptor of protons and its ability to undergo transformations between structurally ordered and disordered states. However, 1,4-diisopropyl-1,4-diazonia-bicyclo[2.2.2]octane dibromide, as a potential bridging ligand, has rarely been studied with respect to its coordination chemistry. Taking all these issues into consideration, as a continuation of our systematic studies of phase transitions,8 we report herein two new compounds, 1,4-diisopropyl-1,4-diazonia-bicyclo[2.2.2]octane tetra-chlorobromo-manganese(II) (1) and 1,4-diisopropyl-1,4-diazonia-bicyclo[2.2.2]octane tetra-chlorobromo-cobalt(II) (2), that have reversible structural phase transition properties in the presence of a novel ligand, and characterize them by variable-temperature single-crystal X-ray diffraction, differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA).
Experimental
Materials and measurements
All reagent-grade chemicals and solvents were obtained from commercial sources and used without further purification. Infrared spectra of compounds 1 and 2 were recorded on a SHIMADZU IR prestige-21 FTIR-8400S spectrometer in the range of 4000–500 cm−1 with samples in the form of potassium bromide pellets (Fig. S1 and S2, ESI†). X-ray powder diffraction data were collected using a Siemens D5005 diffractometer with Cu-Kα radiation (λ = 1.5418 Å). Powder X-ray diffraction (PXRD) patterns of 1 and 2 (Fig. S3 and S4, ESI†) at room temperature matched very well those simulated from the single-crystal structures, confirming the phase purity of 1 and 2 respectively. Elemental analyses were taken on a PerkinElmer 240C elemental analyzer. Thermogravimetric analyses (TGA) of 1 and 2 were conducted on a NETZSCH TG 209 F3 thermogravimeter with a heating rate of 10 K min−1 in a N2 atmosphere (Fig. S5 and S6, ESI†).
Synthesis of 1
Manganese chloride tetrahydrate (1.97 g, 10 mmol) and 1,4-diisopropyl-1,4-diazonia-bicyclo[2.2.2]octane dibromide (3.6 g, 10 mmol) were mixed in aqueous solution (30 ml) (Scheme 1). After being stirred for 20 min in air, the reaction solution was evaporated slowly at room temperature for 3 days, and pale yellow rod-like crystals were obtained in 58% yield (based on manganese chloride). IR spectra of compound 1: 3393(s), 3017(s), 2978(m), 1633(m), 1478(s), 1407(s), 1328(m), 1171(m), 1117(s), 1061(m), 989(w), 893(m), 851(s), 830(s), 815(m), 665(w), 589(w), 555(w). Anal. (%) calcd for C12H26BrCl3MnN2: C, 32.79; H, 5.96; N, 6.37; found: C, 32.61; H, 5.85; N, 6.28.
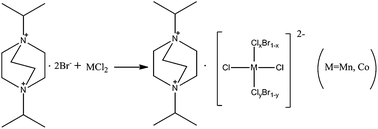 |
| | Scheme 1 Synthesis of compound 1 and 2. | |
Synthesis of 2
Cobalt(II) chloride hexahydrate (2.38 g, 10 mmol) and 1,4-diisopropyl-1,4-diazonia-bicyclo[2.2.2]octane dibromide (3.6 g, 10 mmol) were mixed in aqueous solution (30 ml) (Scheme 1). After being stirred for 20 min in air, the reaction solution was evaporated slowly at room temperature for 5 days, and blue rod-like crystals were obtained in 55% yield (based on cobalt(II) chloride). IR spectra of compound 1: 3439(s), 3016(s), 2975(m), 1638(m), 1478(s), 1402(s), 1326(m), 1174(m), 1110(s), 1063(m), 1032(w), 970(w), 894(m), 861(s), 827(s), 798(w), 683(w), 585(w), 550(w). Anal. (%) calcd for C48H104Br5Cl11Co4N8: C, 31.70; H, 5.76; N, 6.16; found: C, 31.61; H, 5.64; N, 6.08.
Single-crystal X-ray diffraction measurements
Single-crystal X-ray data of compounds 1 and 2 were performed on a Bruker SMART-APEX II CCD with Mo-Kα radiation (λ = 0.71073 Å). A pale yellow rod-like crystal of 1 with approximate dimensions of 0.40 × 0.40 × 0.20 mm was used in data collection at 296 and 150 K respectively. And a blue rod-like crystal of 2 with approximate dimensions of 0.30 × 0.30 × 0.20 mm was similarly used in data collection at 296 and 150 K. Data processing including empirical absorption correction was performed using SADABS. The structures of 1 and 2 were solved by direct methods and refined by using the full-matrix method based on F2 in the SHELXTL software package.9 Non-H atoms were refined anisotropically using all reflections with I > 2σ(I). All H atoms were found in the difference maps. However, carbon-bonded H atoms were added geometrically and refined using the riding model with Uiso = 1.2 Ueq. Asymmetric units and packing views were drawn with DIAMOND (Brandenburg and Putz, 2005). Distances and angles between some atoms were calculated using DIAMOND and other calculations were carried out using SHELXTL. Crystallographic data and structure refinement statistics of 1 and 2 at 296 K and 150 K are listed in Table 1.
Table 1 Crystallographic data for 1 and 2 at RTP and LTP
| |
1 (296 K) |
1 (150 K) |
2 (296 K) |
2 (150 K) |
| Empirical formula |
C12H26BrCl3MnN2 |
C12H26BrCl3MnN2 |
C48H104Br5Cl11Co4N8 |
C48H104Br5Cl11Co4N8 |
| Formula weight |
439.55 |
439.55 |
1818.61 |
1818.61 |
| Crystal system |
Orthorhombic |
Orthorhombic |
Orthorhombic |
Orthorhombic |
| Space group |
Pnma |
P212121 |
Pnma |
P212121 |
| a (Å) |
13.778(3) |
13.6378(11) |
13.7347(15) |
13.5796(9) |
| b (Å) |
10.235(2) |
10.1863(8) |
10.1454(11) |
10.0792(7) |
| c (Å) |
13.238(3) |
13.1566(11) |
13.1412(14) |
13.0655(9) |
| α (°) |
90 |
90 |
90 |
90 |
| β (°) |
90 |
90 |
90 |
90 |
| γ (°) |
90 |
90 |
90 |
90 |
| V (Å3) |
1866.7(7) |
1827.7(3) |
1831.1(3) |
1788.3(2) |
| Z |
4 |
4 |
1 |
1 |
| Dc (g m−3) |
1.564 |
1.597 |
1.649 |
1.689 |
| μ (mm−1) |
3.267 |
3.337 |
4.056 |
4.153 |
| F (000) |
892 |
892 |
918 |
918 |
| θ range [°] |
2.13 to 25.68 |
2.15 to 25.67 |
2.14 to 25.67 |
2.16 to 25.67 |
| Collected reflections |
13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 530 530 |
13842 |
13326 |
13624 |
| Unique reflections |
1879 |
3461 |
1840 |
3389 |
| R1, wR2 [I > 2σ(I)] |
0.0510, 0.1677 |
0.0314, 0.1068 |
0.0687, 0.2379 |
0.0402, 0.1076 |
| R1, wR2 [all data] |
0.0642, 0.1835 |
0.0330, 0.1118 |
0.0815, 0.2653 |
0.0417, 0.1119 |
| GOF |
1.490 |
0.985 |
1.166 |
1.074 |
| Largest peak and hole (e Å−3) |
1.213, −1.073 |
0.649, −0.786 |
1.573, −2.674 |
1.276, −1.406 |
DSC measurement
DSC runs of crystal 1 (17.5 mg) and 2 (24.26 mg) were recorded using a NETZSCH DSC 200 F3 instrument from 280 to 180 K and 270 to 190 K respectively with a heating rate of 15 K min−1 and 10 K min−1 respectively under nitrogen at atmospheric pressure in aluminum crucibles.
Results and discussion
DSC
Differential scanning calorimetry (DSC) is one of the thermodynamic methods used to detect the dependence of reversible phase transition on temperature. Heating a crystalline sample of 1 showed a main endothermic peak at 249.6 K, and cooling the sample yielded a main exothermic peak at 245.2 K (Fig. 1), with the enthalpy changes of ΔHh = 0.6832 J g−1 and ΔHc = 0.5646 J g−1, respectively. The step-like anomalies of the curves at these two main peaks and the thermal hysteresis of 4.4 K reveal that the sample of 1 had undergone a reversible phase transition. Similarly, heating a crystalline sample of 2 yielded a main endothermic peak at 227.8 K, and cooling the sample yielded and a main exothermic peak at 222.3 K (Fig. 2), with the enthalpy changes of ΔHh = 0.4253 J g−1 and ΔHc = 0.3964 J g−1, respectively. The step-like anomalies and the thermal hysteresis of 5.5 K reveal that a reversible phase transition occurred for 2 as well.
 |
| | Fig. 1 DSC curves of 1 obtained in a heating–cooling mode. | |
 |
| | Fig. 2 DSC curves of 2 obtained in a heating–cooling mode. | |
Crystal structure of 1
The phase transition of 1 was further confirmed by determining its crystal structures at room temperature (296 K) and at a low temperature (150 K), respectively (Fig. 3a and b). The crystal structure of 1 at room temperature (296 K) was shown to be orthorhombic with a centrosymmetric space group of Pnma and the point group D2h. When the temperature was decreased to 150 K, 1 transformed to a different orthorhombic crystal structure with the chiral space group P212121 and point group D2. Thereafter the crystal structures of 1 were compared at 296 K and 150 K. At room temperature (296 K), the crystals were determined to be of the orthorhombic space group Pnma (no. 62). When cooled to 150 K, the crystals transformed into the orthorhombic space group P212121 (no. 19). Visual inspection showed no obvious changes between the crystals at the two temperatures. The molecular volume decreased from 1866.7(7) Å3 in the room-temperature phase (RTP) to 1827.7(3) Å3 in the low-temperature phase (LTP).
 |
| | Fig. 3 View of the asymmetric unit of 1 with atomic numbering scheme at (a) 296 K and (b) 150 K. | |
In the room-temperature phase (RTP, 296 K), the structure of 1,4-diisopropyl-1,4-diazonia-bicycle[2.2.2]octane tetra-chlorobromo-manganese(II) consists of 1,4-diisopropyl-1,4-diazonia-bicyclo[2.2.2]octane dications and tetrahedral MnBrCl3 anions (Fig. 3a). The Mn(II) ion adopts a distorted tetrahedral geometry, in which it coordinates Cl atoms at two of the four liganding positions, and either Br or Cl at the other two positions. That is, these latter two positions exhibit substitutional disorder of halogen atoms in the asymmetric unit, with a 0.5 occupancy for Cl and 0.5 occupancy for Br. In this RTP asymmetric unit, there are 9 non-hydrogen atoms ((Br3\Cl3), (Br1\Cl1), Mn1, C7, N2, C4, C3, N1 and C2) located in a mirror plane, each with an occupancy factor of 0.5, and other non-H atoms away from the above mirror plane can be produced by a (x, −y + 3/2, z) symmetry transformation. It is notable that the lengths of the Mn–Br and Mn–Cl bonds are strikingly different (Table S1†), that is, the bond length between Mn1 and the substitutionally disordered Br1 is 2.452(3) Å and the bond length of Mn1 with the other substitutionally disordered Br (Br3) is 2.462(12) Å. The distance between Mn1 and Cl2 is 2.3706(10) Å, and this bond length is obviously shorter than the 2.459(18) Å bond length of Mn1 with the substitutionally disordered Cl1, and it is a little longer than the 2.332(12) Å bond length of Mn1 with the substitutionally disordered Cl3.
In the lower-temperature phase (LTP, 150 K) crystal structure, the Mn(II) ions still adopt the distorted tetrahedral geometry (Fig. 3b). However, the lengths of the four Mn–Cl bonds, i.e., Mn1–Cl1 = 2.390(5) Å, Mn1–Cl2 = 2.3909(8) Å, Mn1–Cl3 = 2.3536(10) Å and Mn1–Cl4 = 2.448(13), are all different from the corresponding lengths in the high-temperature structure (Table S2†). The lengths of the two Mn–Br bonds are 2.436(4) Å (Mn1–Br1) and 2.476(2) Å (Mn1–Br4), respectively. Meanwhile, the conformations of the rings of the dabco ligand show some difference between the two temperature phases. The values of the N–C–C–N torsion angles are 0°, 0°, 0° in the RTP and 23.3(4)°, 22.7(4)°, 24.9(4)° in the LTP. In the LTP unit cell (Fig. 4b), 9 non-hydrogen atoms ((Br3\Cl3), (Br1\Cl1), Mn1, C7, N2, C4, C3, N1 and C2) apparently deviate from the crystallographic mirror plane.
 |
| | Fig. 4 Unit cell packing diagrams of 1 at (a) 296 K and (b) 150 K. | |
X-ray crystal structures of 1 were also determined at 290 K, 280 K, 270 K, 260 K, 250 K, 240 K, 230 K, 220 K, 210 K, 200 K, 190 K, 180 K and 170 K. The cell parameters of 1 measured at the LTP slightly differ from those measured at RTP, as is shown in Fig. 5.
 |
| | Fig. 5 Temperature dependence of unit-cell length parameters of 1. | |
TGA of 1
To study the thermal stability of 1 and to further confirm its molecular formula, thermogravimetric (TG) analysis was performed in N2 on a sample of complex 1 in the range of 25–1000 °C (Fig. S5†). The TGA curve indicates that compound 1 exhibits relatively high thermal stability. The decomposition temperature was measured to be about 263.8 °C, corresponding to the release of organic ligands. Upon increasing the temperature, the structure of 1 began to collapse.
Crystal structure of 2
The phase transition of 2 was further confirmed by determining crystal structures at room temperature (296 K) and a low temperature phase (150 K) (Fig. 6a and b). The crystal structure of 2 at room temperature (296 K) was shown to be orthorhombic with a centrosymmetric space group of Pnma and the point group D2h. When the temperature was decreased to 150 K, 2 transformed to another orthorhombic crystal structure with the chiral space group P212121 and the point group D2. Thereafter the crystal structures of 2 were compared at 296 K and 150 K. At room temperature (296 K), the crystals were observed to be in the orthorhombic space group Pnma (no. 62). When cooled to 150 K, the crystals transformed to the orthorhombic space group P212121 (no. 19). Visual inspection showed no obvious changes between the crystals at the two temperatures. The molecular volume decreased from 1831.1(3) Å3 in the RTP to 1788.3(2) Å3 in the LTP.
 |
| | Fig. 6 View of the asymmetric unit of 2 with atomic numbering scheme at (a) 296 K and (b) 150 K. | |
In the room-temperature phase (RTP, 296 K), the structure of 1,4-diisopropyl-1,4-diazonia-bicyclo[2.2.2]octane tetra-chlorobromo-cobalt(II) consists of 1,4-diisopropyl-1,4-diazonia-bicyclo[2.2.2]octane dications and tetrahedral CoBr1.25Cl2.75 anions (Fig. 6a). The Co(II) ion adopts a distorted tetrahedral geometry in which it coordinates two equivalent Cl atoms and one Br atom at three of the four ligand positions and either Br or Cl at the remaining position. That is, this position exhibits substitutional disorder of halogen atoms in the asymmetric unit, with a 0.75 occupancy for Cl and 0.25 occupancy for Br. In the asymmetric unit of the RTP structure, there are 9 non-hydrogen atoms (Br2, (Br3\Cl3), Co1, C7, N2, C6, C4, N1 and C2) located in a mirror plane, each with an occupancy factor of 0.5, and other non-H atoms away from the above mirror plane can be produced by a (x, −y + 1/2, z) symmetry transformation. It is notable that the lengths of the Co–Br and Co–Cl bonds are strikingly different (Table S3†), that is, the bond length between Co1 and the substitutionally disordered Br3 is 2.39(3) Å and the bond length of Co1 with the other Br (Br2) is 2.3733(15) Å, which are comparable to the corresponding Mn–Br bond lengths in 1. The distance between Co1 and Cl1 is 2.2855(13) Å, and this bond length is a bit longer than the 2.26(2) Å length of the bond connecting Co1 and the substitutionally disordered Cl3.
In the lower-temperature phase (LTP, 150 K) crystal structure, the Co(II) ions still adopt the distorted tetrahedral geometry (Fig. 6b). However, the lengths of the three Co–Cl bonds, i.e., Co1–Cl1 = 2.2734(14) Å, Co1–Cl2 = 2.3048(12) Å, and Co1–Cl4 = 2.323(12), are all different from the corresponding lengths in the high-temperature structure (Table S4†). The bond lengths of the two Co–Br bonds are 2.3926(8) Å (Co1–Br3) and 2.341(14) Å (Co1–Br4), respectively, which also provide a direct comparison to compound 1. Meanwhile, conformations of the rings of the dabco ligand show some difference between the two temperature phases. The values of the N–C–C–N torsion angles are 0°, 0°, 0° in the RTP and 22.7(6)°, 22.2(6)°, 24.6(6)° in the LTP, respectively. In the LTP unit cell (Fig. 7b), 9 non-hydrogen atoms (Br2, (Br3\Cl3), Co1, C7, N2, C6, C4, N1 and C2) apparently deviate from the crystallographic mirror plane.
 |
| | Fig. 7 Unit cell packing diagrams of 2 at (a) 296 K and (b) 150 K. | |
X-ray crystal structures of 2 were also determined at 290 K, 270 K, 250 K, 230 K, 220 K, 210 K, 200 K, 190 K and 170 K. The cell parameters of 2 measured at LTP are slightly different from those measured at RTP, as was observed for 1 (Fig. S6†).
TGA of 2
To study the thermal stability of 2 and to further confirm its molecular formula, thermogravimetric (TG) analysis was performed in N2 on a sample of complex 2 in the range of 25–1000 °C (Fig. S7†). The TGA curve indicates that compound 2 exhibits relatively high thermal stability, as for compound 1. The decomposition temperature was measured to be about 230 °C, corresponding to the release of organic ligands. Upon increasing the temperature, the structure of 2 began to collapse.
Compounds 1 and 2 underwent a phase transition (at ca. T = 245.2 K and ca. T = 222.3 K respectively), from a room-temperature phase with a space group of Pnma to a low-temperature phase with a chiral space group P212121, respectively. During the transition from the RTP to the LTP, Landau symmetry breaking occurred with a pure GPT (mmm–222).10 In other words, the eight symmetry elements (E, C2, 2C2′, i, σh, 2σv) at RTP were halved into four symmetry elements (E, C2, 2C2′) at LTP owing to the loss of i, σh and 2σv. The mirror symmetry in [010] was broken and the inversion center disappeared. The 2-fold screw axis remained unchanged, leading to the final low-temperature space group P212121. Note that the number of the symmetry operations in Pnma decreased by half from 8 [(1) 1; (2) 2(0, 0, 1/2) 1/4, 0, z; (3) 2(0, 1/2, 0) 0, y, 0; (4) 2(1/2, 0, 0) x, 1/4, 1/4; (5) ![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0, 0, 0; (6) a x, y, 1/4; (7) m x, 1/4, z; (8) n(0, 1/2, 1/2) 1/4, y, z] to 4 [(1) 1; (2) 2(0, 0, 1/2) 1/4, 0, z; (3) 2(0, 1/2, 0) 0, y, 1/4; (4) 2(1/2, 0, 0) x, 1/4, 0] in P212121, in perfect agreement with the symmetry breaking analysis (Fig. 8). A strategy similar to that described in the current work that combines engineering crystals by varying the temperature with Landau phase transition theory may also find application in ferroelastics design.
0, 0, 0; (6) a x, y, 1/4; (7) m x, 1/4, z; (8) n(0, 1/2, 1/2) 1/4, y, z] to 4 [(1) 1; (2) 2(0, 0, 1/2) 1/4, 0, z; (3) 2(0, 1/2, 0) 0, y, 1/4; (4) 2(1/2, 0, 0) x, 1/4, 0] in P212121, in perfect agreement with the symmetry breaking analysis (Fig. 8). A strategy similar to that described in the current work that combines engineering crystals by varying the temperature with Landau phase transition theory may also find application in ferroelastics design.
 |
| | Fig. 8 Changes of symmetry operations of 1 and 2 from Pnma at RTP to P212121 at LTP. | |
Conclusions
In summary, DSC, variable-temperature structural analysis and TGA revealed that compounds 1 and 2 underwent a reversible phase transition, with a discontinuity at ca. 245.2 K and ca. 222.3 K, respectively. Crystal structures of 1 and 2 obtained at room temperature and a low temperature revealed that they reversibly transformed from an RTP space group of Pnma to an LTP space group of P212121. During the transition from the RTP to LTP, Landau symmetry breaking occurred with a pure GPT (mmm–222) in both compounds 1 and 2. Owing to the ordering of the twisting motions of the 1,4-diisopropyl-1,4-diazonia-bicyclo[2.2.2]octane ion, the conformations of the rings of dabco evidently differed between RTP and LTP phases. The ordering of the twisting motions of the dabco ring probably drove the phase transition.
Acknowledgements
This work was funded by the National Natural Science Foundation of China (Grant no. 21201087), JiangSu Province NSF BK20131244, the Foundation of Jiangsu Educational Committee (11KJB150004) and sponsored by Qing Lan Project of Jiangsu province and Jiangsu Overseas Research & Training Program for University Prominent Young & Middle-aged Teacher and Presidents, the Innovation Program of Graduate Students in Jiangsu Province (SJZZ_0142).
References
-
(a) D. Lencer, M. Salinga and M. Wuttig, Adv. Mater., 2011, 23, 2030–2058 CrossRef CAS PubMed;
(b) M. Salinga and M. Wuttig, Science, 2011, 332, 543 CrossRef CAS PubMed;
(c) H. Zheng, J. B. Rivest, T. A. Miller, B. Sadtler, A. Lindenberg, M. F. Toney, L.-W. Wang, C. Kisielowski and A. P. Alivisatos, Science, 2011, 333, 206–209 CrossRef CAS PubMed;
(d) J. F. Scott and C. A. P. Dearaujo, Science, 1989, 246, 1400–1405 CAS.
-
(a) J. F. Scott, Science, 2007, 315, 954–959 CrossRef CAS PubMed;
(b) S. Horiuchi and Y. Tokura, Nat. Mater., 2008, 7, 357–366 CrossRef CAS PubMed;
(c) I. R. Evans, J. A. K. Howard and J. S. O. Evans, Cryst. Growth Des., 2008, 8, 1635–1639 CrossRef;
(d) G.-C. Xu, X.-M. Ma, L. Zhang, Z.-M. Wang and S. Gao, J. Am. Chem. Soc., 2010, 132, 9588–9590 CrossRef CAS PubMed.
-
(a) M. Szafrański and M. Jarek, CrystEngComm, 2013, 15, 4617–4623 RSC;
(b) Z.-H. Sun, J.-H. Luo, T.-L. Chen, L.-N. Li, R.-G. Xiong, M.-L. Tong and M.-C. Hong, Adv. Funct. Mater., 2012, 22, 4855–4861 CrossRef CAS;
(c) Q. Ye, T. Akutagawa, H.-Y. Ye, T. Hang, J.-Z. Ge, R.-G. Xiong, S.-i. Noro and T. Nakamura, CrystEngComm, 2011, 13, 6185–6191 RSC.
-
(a) Z.-H. Sun, X.-Q. Wang, J.-H. Luo, S.-Q. Zhang, D.-Q. Yuan and M.-C. Hong, J. Mater. Chem. C, 2013, 1, 2561–2567 RSC;
(b) Y. Zhang, H.-Y. Ye, D.-W. Fu and R.-G. Xiong, Angew. Chem., 2014, 126, 2146–2150 CrossRef.
- H. Zhang, X.-M. Wang, K.-C. Zhang and B. K. Teo, Coord. Chem. Rev., 1999, 183, 157–195 CrossRef CAS.
-
(a) H.-Y. Ye, H.-L. Cai, J.-Z. Ge and R.-G. Xiong, Inorg. Chem. Commun., 2012, 17, 159–162 CrossRef CAS PubMed;
(b) Y. Zhang, W. Zhang, S.-H. Li, Q. Ye, H.-L. Cai, F. Deng, R.-G. Xiong and S. P. D. Huang, J. Am. Chem. Soc., 2012, 134, 11044–11049 CrossRef CAS PubMed.
-
(a) Y. Zhang, W.-Q. Liao, H.-Y. Ye, D.-W. Fu and R.-G. Xiong, Cryst. Growth Des., 2013, 13, 4025–4030 CrossRef CAS;
(b) W. Zhang, H.-Y. Ye, H.-L. Cai, J.-Z. Ge, R.-G. Xiong and S.-D. Huang, J. Am. Chem. Soc., 2010, 132, 7300–7302 CrossRef CAS PubMed;
(c) H.-Y. Ye, J.-Z. Ge, F. Chen and R.-G. Xiong, CrystEngComm, 2010, 12, 1705–1708 RSC.
-
(a) L.-Z. Chen, D.-D. Huang, J.-Z. Ge and F.-M. Wang, CrystEngComm, 2014, 16, 2944–2949 RSC;
(b) L.-Z. Chen, H. Zhao, J.-Z. Ge, R.-G. Xiong and H.-W. Hu, Cryst. Growth Des., 2009, 9, 3828–3831 CrossRef CAS;
(c) H.-Y. Ye, L.-Z. Chen and R.-G. Xiong, Acta
Crystallogr., Sect. B: Struct. Sci., 2010, 66, 387–395 CAS.
-
(a) G. M. Sheldrick, SHELXL-97, Program for Crystal Structure Solution, University of Gottingen, Germany, 1997 Search PubMed;
(b) G. M. Sheldrick, SHELXS-97, Program for Crystal Structure Refinement, University of Gottingen, Germany, 1997 Search PubMed.
-
(a) M. J. Tellot, A. López-Echarrit, J. Zubillagat, I. Ruiz-Lamea, F. J. Zúñigat, G. Madariaga and A. Gómez-Cuevast, J. Phys.: Condens. Matter, 1994, 6, 6751–6760 CrossRef;
(b) A. Gómez-Cuevast, J. M. Pérez Mato, M. J. Tello, G. Madariaga, J. Fernández and L. Echarri, Phys. Rev. B: Condens. Matter Mater. Phys., 1984, 29, 2655–2663 CrossRef;
(c) S. Prosandeev, I. A. Kornev and L. Bellaiche, Phys. Rev. Lett., 2011, 107, 117602 CrossRef CAS;
(d) S. Hirotsu, J. Phys. C: Solid State Phys., 1975, 8, L12–L16 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: IR, PXRD and TGA of compounds 1 and 2. CCDC 972354, 972355, 1026402 and 1026403. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ra12690d |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 








