
UV-vis and EPR spectroelectrochemical investigations of triarylamine functionalized arylene bisimides†
Sandra Pluczyka,
Pawel Zassowskia,
Renata Rybakiewiczb,
Renata Wielgoszb,
Malgorzata Zagorskab,
Mieczyslaw Lapkowski*ac and
Adam Pron*b
aSilesian University of Technology, Faculty of Chemistry, Strzody 9, 44-100 Gliwice, Poland. E-mail: mieczyslaw.lapkowski@polsl.pl
bFaculty of Chemistry, Warsaw University of Technology, Noakowskiego 3, 00-664 Warsaw, Poland
cCentre of Polymer and Carbon Materials, Polish Academy of Sciences, M. Curie-Sklodowska 34, 41-819 Zabrze, Poland
First published on 19th December 2014
Abstract
Four arylene bisimides N-substituted with triarylamine and three bisimides core-functionalized with the same substituent were studied by cyclic voltammetry, UV-vis and EPR spectroelectrochemistry. All the investigated compounds showed ambipolar behaviour manifested by their quasi-reversible reduction to radical anions and quasi-reversible oxidation to radical cations. The presence of stable radical anions and radical cations was confirmed by EPR spectroelectrochemical experiments. Formation of the radical anions resulted in bleaching of the bisimide UV-vis bands with simultaneous hypsochromic shift of the charge transfer (CT) band and appearance of the radical anion peaks, the bands originating from the triarylamine remaining essentially unchanged. Electrochemical generation of radical cations resulted in turn in bleaching of the triarylamine band accompanied by a hypsochromic shift of the CT band and with the appearance of the radical cation bands at higher wavelengths, the bisimide bands remained essentially intact.
Introduction
Functionalized arylene bisimides have been used for many years as different types of dyes.1 More recently, they have been applied as active layers in n-channel organic field effect transistors (OFETs)2–8 or as building blocks in photochromic and electrochemically-active metal–organic frameworks.9–13 One of the advantages of these organic semiconductors is the possibility of tuning their electron affinity (EA) through core or imide nitrogen functionalization with appropriate electron accepting (donating) groups.5,14–18Low and high molecular mass triarylamines are in turn very suitable for the fabrication of active layers in p-channel FETs or as hole conducting materials, in general (see for example19 and references therein) or as electrochromic20 and electrofluorochromic21 materials. In their oxidized states they also frequently serve as spin-carrying units in high spin organic materials due to good stability of their radical cation form which in molecules of appropriate topology facilitates ferromagnetic spin coupling.22
Molecules (macromolecules) combining triarylamine and arylene bisimide moieties connected either directly or through appropriate linkers have been designed and studied in the past decade as components of electrochromic materials and/or materials showing volatile and non-volatile memory.23–29 In addition many compounds of this family exhibit ambipolar properties which are scarce and still greatly in demand. Several solution processable arylene bisimides N- or core-substituted with triarylamines have been synthesized.17,30,31 In addition to being attractive organic semiconductors, yielding ambipolar OFETs operating in air,17,31 they show interesting and reversible electrochemical behaviour.32
In this work we describe the results of UV-vis and EPR spectroelectrochemical investigations of arylene bisimides differently substituted with triarylamine. No such data have ever been reported and their publication is interesting because they form stable radical ions both in their oxidized and reduced forms, whose electronic structure is dependent on the size of the bisimide core as well as on the number and positions of triarylamine substituents.
Experimental
Synthesis
The preparation procedures of already published bisimides (B1, B2 and B3) can be found in the ESI of ref. 30 whereas that of B4 in ref. 31 and B5 in ref. 32. The detailed synthetic pathways leading to B6 and B7, together with their spectroscopic characterization, can be found in the ESI† of this paper.Cyclic voltammetry
Cyclic voltammograms of the synthesized compounds were registered using an Autolab potentiostat (EcoChemie) in the electrolytic medium consisting of the studied bisimide dissolved in 0.1 M dichloromethane solution of Bu4NBF4. The measurements were performed in an inert atmosphere, using a platinum working electrode of the surface area of 3 mm2, a platinum wire counter electrode and an Ag/0.1 M Ag+ reference electrode consisting of an Ag wire immersed in 0.1 M solution of AgNO3 in acetonitrile.UV-vis and EPR spectroelectrochemistry
The spectroelectrochemical cell used in the UV-vis spectroelectrochemical investigations was a modification of cells described in ref. 33–35. In all experiments 10−4 M solution of a given bisimide was prepared in 0.1 M Bu4NPF6/dichloromethane electrolyte. The spectra were recorded in a 2 mm thick cuvette using an UV-vis Hewlett Packard 8453 spectrometer. The platinum mesh was used as a working electrode, a platinum spiral as an auxiliary electrode and a silver wire as a pseudo-reference electrode. In the case of solid state spectroelectrochemistry, thin films of a given compound were deposited on an ITO electrode by casting from solution. In this case, dichloromethane was replaced by acetonitrile.Electrochemical cells of several different geometries can be envisioned for EPR spectroelectrochemical investigations,34 the majority of them being flat.36,37 In this research a cylindrical cell was used, similar to that described in ref. 38 and identical to the cell reported in ref. 39. In particular, EPR measurements were carried out in 10−3 M solution of an investigated bisimide in 0.1 M Bu4NPF6/dichloromethane electrolyte using a cylindrical custom – made cell equipped with a platinum wire working electrode, an auxiliary electrode in a form of platinum spiral and a silver wire as a pseudo – reference electrode. The pseudo-reference electrode potential was checked after each experiment by recording a cyclic voltammogram of ferrocene added to the same electrolyte solution. The experiments were performed using a JEOL JES-FA 200, X-band CW-EPR spectrometer operating at 100 kHz field modulation. EPR spectra of the electrochemically generated radicals were fitted using WinSim software40 G-factor was estimated by comparison with JEOL spectrometer internal standard (Mn(II) salt in quartz tube).
DFT/TDDFT calculations
DFT/TDDFT calculations have been carried with B3LYP (ref. 41–43) hybrid functional with 6-31G(d) basis set. Ground state geometry was optimized without symmetry constraints to a local minimum, which was followed by frequency calculations. Alkyl chains were cut to the methyl group, in order to speed up the calculations. No imaginary frequencies were detected, which proved that obtained geometry was a local minimum. All calculations were carried out with a polarizable continuum model,44 using dichloromethane as a solvent in order to simulate the solution effects. All calculations were carried out with Gaussian 09 software.45 Input files and plots were prepared with Gabedit software.46Results and discussion
The investigated compounds are depicted in Chart 1. They can be divided into two groups, imide nitrogen – (B1, B2, B3, B4) and core – (B5, B6, B7) substituted derivatives. | ||
| Chart 1 Chemical formulae of triarylamine-substituted arylene bisimides studied. | ||
Cyclic voltammetry
Representative voltammograms of N- and core-substituted arylene bisimides (B2 and B5) are shown in Fig. 1. Voltammograms of the remaining compounds can be found in Fig. S1 of ESI.† Redox potentials of all studied compounds, derived from the cyclic voltammetry data, are listed in Tables 1 and 2. | ||
| Fig. 1 Cyclic voltammograms of: B2 (a) and B5 (b). Concentration of B2: 1 × 10−3 M and B5: 5 × 10−4 M; electrolyte 0.1 M Bu4NBF4 in CH2Cl2; scan rate 50 mV s−1. | ||
| Compound | Ered1 (V) | Eox1 (V) | 1/2(Ered1 + Eox1) (V) | Ered1 (onset) (V) | Ered2 (V) | Eox2 (V) |
|---|---|---|---|---|---|---|
| B1 | −1.32 | −1.19 | −1.26 | −1.16 | — | — |
| B2 | −1.10 | −1.00 | −1.05 | −0.97 | −1.55 | −1.41 |
| B3 | −1.07 | −0.96 | −1.02 | −0.96 | −1.25 | −1.15 |
| B4 | −1.13 | −1.01 | −1.07 | −1.00 | −1.59 | −1.42 |
| B5 | −1.18 | −1.10 | −1.14 | −1.06 | −1.59 | −1.49 |
| B6 | −1.37 | −1.29 | −1.33 | −1.26 | −1.80 | −1.67 |
| B7 | −1.19 | −1.11 | −1.15 | −1.05 | −1.57 | −1.51 |
| Compound | Eox3 (V) | Ered3 (V) | 1/2(Eox3 + Ered3) (V) | Eox3 (onset) (V) |
|---|---|---|---|---|
| B1 | 0.52 | 0.42 | 0.47 | 0.40 |
| B2 | 0.53 | 0.43 | 0.48 | 0.39 |
| B3 | 0.50 | 0.41 | 0.46 | 0.37 |
| B4 | 0.53 | 0.42 | 0.48 | 0.41 |
| B5 | 0.62 | 0.51 | 0.56 | 0.48 |
| B6 | 0.56 | 0.49 | 0.53 | 0.44 |
| B7 | 0.62 | 0.48 | 0.55 | 0.42 |
The electrochemical features of the registered voltammograms require some comments. In core-substituted bisimides, the peak to peak separation of the forward and reversed waves in the bisimide reduction process is equal to ca. 70 to 80 mV, in line with the corresponding value measured in the same conditions for the ferrocene redox couple. These values exceed by 10 to 20 mV the theoretical ones and reflect higher resistivity of non-aqueous electrolytes used in this research as compared to aqueous ones. In N-substituted compounds this separation is even higher (from 100 V to 130 mV) which is evidently associated with slower kinetics of the electron transfer in these derivatives. For the triarylamine oxidation process the separation of peaks is comparable for both types of compounds (from 70 mV to 110 nm) with the exception of B7 where a larger separation is measured (see Table 2).
The presented voltammograms reflect the mechanism of arylene bisimide reduction.47 For naphthalene and perylene bisimides the two reduction peaks, observed at negative potentials (Ered1 and Ered2) are associated with the successive reduction of the two imide groups to a radical cation in the first stage and to a dianion in the second stage. The first reduction step leads to an increase of the electron density on the arylene core. Thus, the addition of a second electron in the consecutive reduction step is determined by the capability of delocalizing the surplus electron density, imposed on the core during the first reduction step. This capability is strongly dependent on the size of the core. For this reason ΔE = Ered1 − Ered2 is lower for B3 than for B2. In bisimides of even larger cores like in quaterrylene ones, for example, the two reduction peaks merge into one peak of doubled intensity, indicating that both reduction steps described above occur at the same potential.47 For the same reason the reduction of the smallest core bisimide (B1) is limited to the first reduction step only since the surplus electron density in the formed radical anion cannot be efficiently delocalized (see Table 1 and Fig. S1†).
In N-substituted bisimides the potential of the first reduction peak increases from B1 to B3, indicating greater facility of larger cores to accept an extra electron. It should be also pointed out that bisimides of the same core, e.g. naphthalene bisimides, but differently N-substituted with triarylamine (B2 and B4) undergo the first and the second reduction at very similar potentials (Table 1). These findings corroborate the results of DFT calculations presented in ref. 30 and 31 which, for N-substituted bisimides, show orthogonality of the triarylamine phenylene ring attached to imide nitrogen with respect to the plane of the bisimide core and by consequence no conjugation between the substituent and the core. Moreover, they also indicate separation of the frontier orbitals in space with HOMO being located on the triarylamine substituent whereas LUMO – on the bisimide core.
Core-functionalized arylene bisimides are more difficult to reduce. The formal potential of the first redox couple (E10 = 1/2(Ered1 + Eox1)) of B5 is shifted to lower values by 90 mV as compared to the case of B2 in which the triarylamine substituents are attached to the imide nitrogen atoms. This is consistent with DFT calculations of core-substituted arylene bisimides reported in ref. 17. In N-substituted bisimides no or a negligible electron donating effect of triarylamine is observed due to orthogonality of the bisimide core and the substituent phenyl ring attached to bisimide nitrogen. In the case of core-substituted compounds the electron donating effect of triarylamine is observed, leading to an increase of the LUMO level energy which is manifested by the already mentioned decrease of the first reduction peak potential. This effect is amplified in B6 where a secondary amine group is attached directly to the core in addition to the triarylamine substituent (see Table 1).
The quasi-reversible redox couple at positive potentials corresponds to the oxidation of the triarylamine moiety to a radical cation and its consecutive reduction to the neutral form. Note that for triarylamine disubstituted arylene bisimides only one oxidation peak is registered, similarly like in the case of derivatives with only one triarylamine substituent. This means that the oxidation of both substituents occurs concomitantly at the same potential. This is additionally corroborated by the integration of the peaks corresponding to the formation of the radical anion and the radical cation, respectively. In the case of N-substituted derivatives (B1–B4), the formal potential of this redox couple (E30 = 1/2(Eox3 + Ered3)) is essentially independent of the core-size, clearly corroborating previous DFT calculations again indicating orthogonality of the core and the substituent and the lack of conjugation (see Table 2). Triarylamine groups in core-substituted derivatives (B5–B7) are more difficult to oxidize than in the N-substituted ones (B1–B4), reflecting the electron accepting effect of the bisimide moiety on the triarylamine substituent, again in accordance with previous DFT calculations.17,31
UV-vis spectroscopy
UV-vis spectra of N-substituted arylene bisimides (B1–B4) have some common features: (i) a strong band in the vicinity of 300 nm characteristic of the triarylamine substituent. The position of this band is independent of the size of the bisimide core (see Table 3): (ii) a band at higher wavelengths, corresponding to the π–π* transition in the bisimide core. Its position and its relative intensity with respect to the triarylamine band are dependent on the core size. In the case of B1 (pyromellitic bisimide) this band strongly overlaps with the triarylamine band. For B2–B4 it shows a clear vibrational structure with the peak originating from the 0–0 transition being the most intensive. A representative spectrum of this group of bisimides (B2) is shown in Fig. 2a whereas the spectra of other bisimides of this group can be found in Fig. S2 of ESI.† All these spectra can be considered as a superposition the spectrum of “free” triarylamine and that of the corresponding arylene bisimide containing non-chromophoric N-substituents, for example alkyl groups.48 This again supports previous DFT calculations which indicate no conjugation between the substituent and the core.30,31| Compound state | λmax [nm] |
|---|---|
| a Thin solid film spectra. | |
| B1 (neutral) | 309; 368 |
| B1− (radical anion) | 415; 658; 727 |
| B1+ (radical cation) | 593; 703 |
| B2 (neutral) | 299; 342 (0–2); 360 (0–1); 381 (0–0) |
| B2− (radical anion) | 273; 401; 478; 609; 699; 780 |
| B22− (dianion) | 273; 401; 426; 609 |
| B2+ (radical cation) | 568; 698 |
| B3 (neutral) | 302; 459 (0–2); 491 (0–1); 527 (0–0) |
| B3− (radical anion) | 682 (0–3); 704 (0–1); 714 (0–0); 797; 975 |
| B32− (dianion) | 574 |
| B3+ (radical cation) | 354; 697 |
| B4 (neutral) | 300; 342 (0–2); 360 (0–1); 381 (0–0) |
| B4− (radical anion) | 272; 402; 478; 610; 688; 763 |
| B42− (dianion) | 272; 402; 425; 610 |
| B4+ (radical cation) | 568; 698 |
| B5 (neutral) | 307; 339 (0–2); 381 (0–0); 592 |
| B5− (radical anion) | 487; 534; 705; 785 |
| B52− (dianion) | 412; 630 |
| B5+ (radical cation) | 508; 707 |
| B6 (neutral) | 307; 339 (0–2); 381 (0–0); 592 |
| B6− (radical anion) | 487; 534; 705; 785 |
| B62− (dianion) | 412; 630 |
| B6+ (radical cation) | 502; 535; 684 |
| B7a (neutral) | 310; 349; 620 |
| B7−a (radical anion) | 303; 485; 530; 703; 785 |
| B7+a (radical cation) | 749 |
 | ||
| Fig. 2 Solution UV-vis spectra of neutral B2 (a) and B5 (b) (concentration: 1 × 10−4 M in CH2Cl2). | ||
The spectra of core-substituted bisimides are significantly different. A representative spectrum of this group of bisimides (B5) is shown in Fig. 2b. It shows a new broad band, nonexistent in the spectra of N-substituted bisimides, with a maximum at 592 nm, whose presence is associated with a charge transfer (CT)-type transition between the triarylamine and the bisimide core which are directly connected.49–52 The band of vibronic nature, originating from the π–π* transition of the bisimide core, is significantly altered, as compared to the case of N-substituted bisimides. The lines corresponding to particular transitions are broadened: the 0–2 transition at 339 nm becomes dominant, with the 0–1 one present as a shoulder and the 0–0 transition giving rise to a weak peak at 381 nm. The peak characteristic of the triarylamine moiety is bathochromically shifted to 309 nm, i.e. by ca. 10 nm as compared to the corresponding band in N-substituted bisimides. All these changes reflect the presence of donor–acceptor interactions in core-functionalized bisimides, predicted by DFT calculations.17 These interactions not only influence the spectral properties of these compounds but also alter their oxidation and reduction potentials (vide supra). UV-vis spectra of other bisimides of this group can be found in Fig. S3 of ESI.†
Spectroelectrochemistry in the reduction mode
In Fig. 3 UV-vis-NIR spectra of B2, registered for decreasing working electrode potentials are collected. The first spectroscopic signs of the electrochemical reduction appear at E = −1.00 V vs. Fc/Fc+ which almost perfectly coincides with the onset of the first reduction peak in the cyclic voltammogram (−0.97 V vs. Fc/Fc+, see Table 1). Within the potential range of this voltammetric peak (−1.0 V > E > −1.30 V) the following bands attributable to the radical anion form of B2 appear and grow in intensity: 273 nm; 401 nm, 478 nm, 609 nm, 699 nm reaching the maximum intensity for potentials −1.25 to −1.30 V. They can be unequivocally attributed to the radical anion form of B2 in accordance with previous findings for arylene bisimides functionalized with non-chromophoric N-substituents.9,53 For potentials corresponding to the onset of the second reduction peak (−1.35 V) and a potential close to its maximum (−1.50 V) the above listed bands decrease in intensity and a new peak, attributable to the dianion form appears at 426 nm. Concomitantly, the π–π* band of vibronic character at 381 nm, which is indicative of the neutral form of the bisimide, decreases in intensity over the whole potential range studied.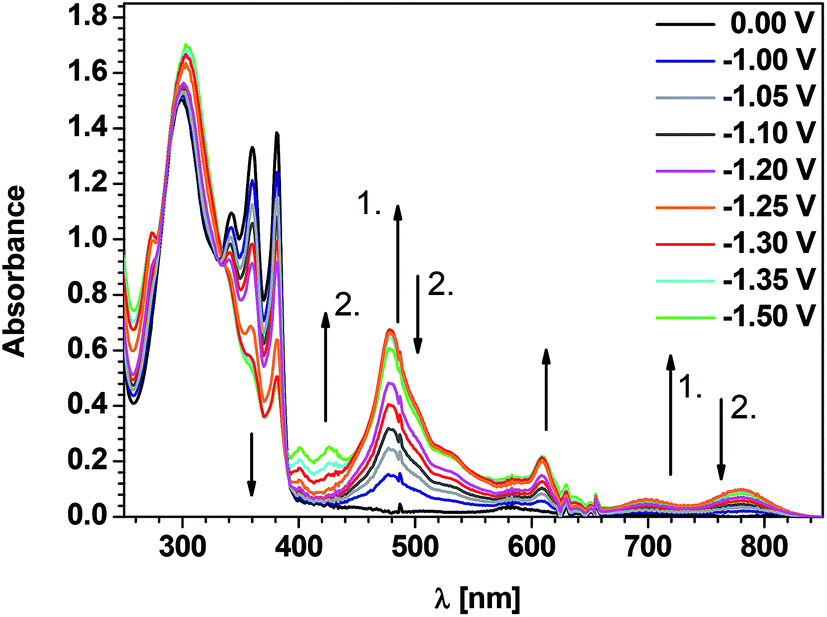 | ||
| Fig. 3 UV-vis spectra of B2 registered for decreasing working electrode potential (concentration: 1 × 10−4 M; electrolyte 0.1 M Bu4NPF6 in CH2Cl2).↑1.– absorption bands which grow during the first step of reduction; ↑2. – absorption bands which grow during the second step of reduction; ↓2. – absorption bands which decrease during the second step of reduction. | ||
The triarylamine band at 299 nm does not change its position with decreasing potential, its intensity however increases which may be caused in part by the changing background of the spectrum. It can therefore be concluded that the reduction of B2 is strictly limited to the bisimide core consistent with the cyclic voltammetry results and previous DFT calculations.30,31 All bisimides of this group behave similarly, although the positions of their π–π* band as well as the bands corresponding to the radical anion and dianion forms are dependent on the bisimide core size (see Table 3 and Fig. S4 in ESI†). Similar behaviour has also been reported for an alternating copolymer of naphthalimide and bis(triarylamine), however in this macromolecular compound the vibronic peaks of the bisimide π–π* transition are significantly broadened. This effect, together with a bathochromic shift of the triarylamine peak causes all these bands to nearly merge.24
B5 i.e. naphthalene bisimide core-substituted with triarylamine is more difficult to reduce than its N-substituted counterpart. Its UV-vis spectrum remains unchanged down to E = −1.10 V vs. Fc/Fc+. At this potential spectroscopic features characteristic of the radical anion state start to appear (see Fig. 4), again in perfect accordance with the onset of the first reduction peak in the cyclic voltammogram of B5 (compare Fig. 1b and 4). In the potentials range covering the first reduction peak in the cyclic voltammogram of B5 (−1.10 V > E > −1.40 V) the band attributed to the π–π* transition in the bisimide core (at 381 nm (0–0) and 339 nm (0–2)) decreases in intensity whereas the triarylamine band at 307 nm increases in intensity. Spectral changes above 400 nm are more complicated and their interpretation requires a substantial theoretical support. In particular, it is important to compare theoretically calculated spectra of the neutral and charged forms of B5. The calculated spectra (at TDDFT/B3LYP/6-31G(d) level of theory) can be found in Fig. S8 of ESI.† The following conclusions can be drawn which are consistent with the experimentally observed spectroelectrochemical behaviour. The CT band at 592 nm in the experimental spectrum undergoes a hypsochromic shift to 484 nm upon the reduction of B5 to a radical anion. A hypsochromic shift of this band is also found in the calculated spectrum. At lower potentials corresponding to the electrochemical generation of dianions (−1.45 V and −1.50 V) this band diminishes whereas two new bands appear at 412 nm and 630 nm and grow in intensity.
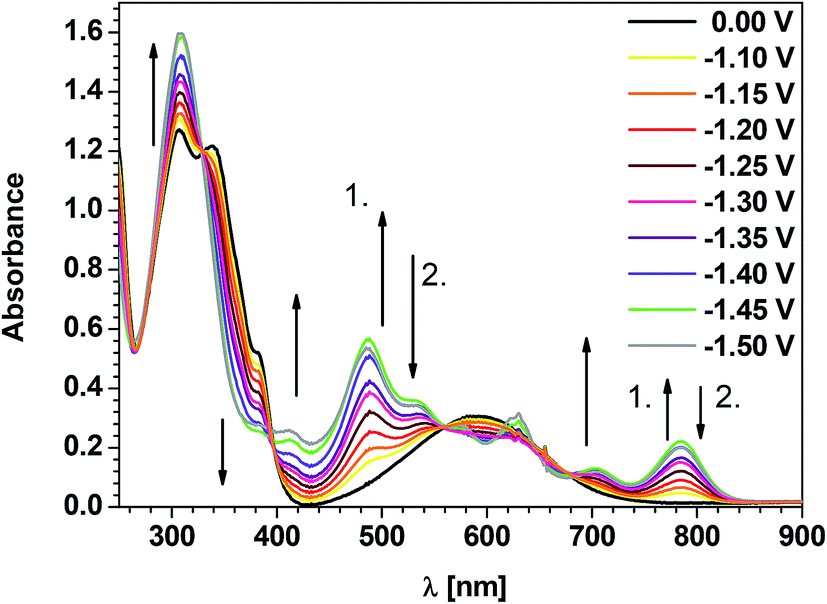 | ||
| Fig. 4 UV-vis spectra of B5 registered for decreasing working electrode potential (concentration: 1 × 10−4 M; electrolyte 0.1 M Bu4NPF6 in CH2Cl2). ↑1. – absorption bands which grow during the first step of reduction; ↓2. – absorption bands which decrease during the second step of reduction. | ||
Similar behaviour is found for B6 (Fig. S5 in ESI†), however in this case clear spectral changes appear at E = −1.30 V vs. Fc/Fc+ consistent with the registered cyclic voltammogram (see Fig. S1 in ESI†). Generally in core substituted bisimides both the π–π* band and the CT band are affected, although the position of the reduction induced-bands depends on the size of the bisimide core.54
Spectroelectrochemical investigations in the solid state were carried out for thin films of the only polymeric bisimide (B7). In Fig. 5 UV-vis-NIR spectra of this polymer, registered in the potentials range covering the first reduction peak, are collected. In the spectrum of neutral B7 the π–π* band of the bisimide core is located at 349 nm and shows no vibrational structure. It strongly overlaps with the triarylamine band of λmax at 310 nm. The CT band is bathochromically shifted to 620 nm as compared to the corresponding band in the spectra of the other two core-functionalized bisimides (B5 and B6). The first reduction-induced spectral changes appear at E = −0.95 V vs. Fc/Fc+ i.e. at a potential ca. 100 mV higher than the potential of the onset of the of the first reduction peak in the cyclic voltammogram registered for a solution of B7 in dichloromethane (see Table 1). Within the potentials range of the first reduction peak (from −1.05 to −1.30 V) the bisimide band bleaches. The triarylamine band increases in intensity and hypsochromically shifts by 7 nm. The CT band undergoes a hypsochromic shift to 485 nm as predicted in the calculations carried out for its low molecular weight analogue (B5). New bands appear at 703 nm and 784 nm.
 | ||
| Fig. 5 UV-vis spectra of B7 registered for decreasing working electrode potential (0.1 M Bu4BF4/CH3CN). ↑1. – absorption bands which grow during the first step of reduction; ↑2. – absorption bands which grow during the second step of reduction; ↓2. – absorption bands which decrease during the second step of reduction. | ||
In all studied bisimides stable radical anions can be electrochemically generated by lowering the working electrode potential to the values corresponding to the first reduction peak. In Fig. 6a–d EPR spectra of N-substituted bisimides are compared. Multi-line, well resolved spectra were observed in almost all cases, indicating that no microaggregation of the bisimide occurs during the electrochemical reduction and rapid reorientation of the formed radical anions takes place which results, in turn, in averaging to zero the anisotropic contributions to the g factor and to hyperfine interactions.
 | ||
| Fig. 6 Experimental and simulated EPR spectra of electrochemically generated radical anions of arylene bisimides N-substituted with triarylamine (1 × 10−3 M; electrolyte 0.1 M Bu4NPF6 in CH2Cl2): (a) B1 (E = −1.2 V); (b) B2 (E = −1.0 V); (c) B3 (E = −1.0 V); (d) B4 (E = −1.0 V). Modulation width in range of 0.01–0.05 G; microwave power 1 mW. Fitting parameters can be found in the ESI (Chart S1†). | ||
B1–B4 give multi-line EPR spectra centered at g in the range of 2.0037–2.0047. The observed patterns can be simulated assuming isotropic hyperfine interactions of the unpaired electron with the nuclei of nitrogen atoms and protons of the bisimide core, there is no evidence of the interactions of the unpaired electron with nitrogen atoms or protons of the triarylamine N-substituent. The lack of such interactions is additionally corroborated by close similarity of the spectra of B2 and B3 to those reported for naphthalene53 and perylene17 bisimides N-substituted with alkyl or oligoether groups as well as the similarity of the spectrum of B4 to that reported for naphthalene bisimide unsymmetrically N-substituted with alkyl and aryl substituents.55 EPR parameters obtained for the radical anion forms of all bisimides studied are presented in Table 4 and in greater detail in Chart S1 of the ESI.†
| Compound | hfcc/G | Linewidth/G | g-factor |
|---|---|---|---|
| B1− | N: 1.18; H: 0.64 | 0.09 | 2.0041 |
| B2− | N: 0.93; H: 1.86 | 0.42 | 2.0037 |
| B3− | N: 0.57; H: 1.75; 0.61 | 0.20 | 2.0039 |
| B4− | N: 0.99; 0.89; H: 1.96; 1.75 | 0.40 | 2.0041 |
| B5− | N: 0.88; H: 1.59; 0.23 | 0.35 | 2.0047 |
| B6− | N: 1.35, 1.05, 1.12; H: 4.04, 1.01, 0.9, 0.7 | 0.40 | 2.0046 |
| B7− | — | — | 2.0039 |
As judged from the coupling constants, in the symmetrically N-substituted bisimides (B1, B2 and B3) the maximum spin density of the unpaired electron is located in the center of the arylene core; in the asymmetric derivative (B4) it is slightly displaced towards this half of the core which is connected to the triarylamine moiety.
EPR spectra of core-functionalized bisimides are shown in Fig. 7 whereas the corresponding spectral parameters are listed in Table 4 and in Chart S1 of the ESI.†
 | ||
| Fig. 7 Experimental and simulated EPR spectra of electrochemically generated radical anions of arylene bisimides core-substituted with triarylamine (1 × 10−3 M; electrolyte 0.1 M Bu4NPF6 in CH2Cl2: (a) B5 (E = −1.1 V); (b) B6 (E = −1.3 V); (c) B7 (E = −1.1 V). Modulation width in range of 0.01–0.05 G; microwave power 1 mW. Fitting parameters can be found in ESI (Chart S1†). | ||
From the set of the coupling constants obtained for B5 i.e. symmetrically core-disubstituted naphthalene bisimide, it can be concluded that similarly as in B2 – its N-substituted counterpart – the maximum spin density of the unpaired electron is located in the center of the aromatic core. B6 is an interesting case since it contains two non-equivalent substituents, one of them being attached to the core via amine-type nitrogen. The electron donating effect of this substituent has a profound effect on the distribution of the unpaired electron spin density whose maximum is displaced to this part of the aromatic core which is the most distant from the amine substituent (note the significantly enhanced coupling constant for B1, Chart S1 in ESI†). A similar phenomenon was previously reported for perylene bisimides core-functionalized with secondary amines.54 Different behaviour was observed for B7. As a macromolecular equivalent of B5, it should exhibit similar EPR spectral parameters. However, only a single broad signal with no hyperfine splitting was recorded. This probably results from the higher molecular weight of this compound, which could lead to aggregation of charged polymer chains. No model was proposed here since many different combinations can be fitted into this spectrum.
To summarize this part of the work, arylene bisimides N- and core-substituted with triarylamine show very interesting electrochemistry in the reduction mode. They undergo quasi-reversible reduction at relatively high potentials yielding radical anions which show excellent stability at room temperature. Moreover, the presence of an additional chromophore (triarylamine), makes the spectroelectrochemical behaviour of these compounds distinctly different from that reported for arylene bisimides which do not contain chromophoric substituents.
Spectroelectrochemistry in the oxidation mode
Low and high molecular weight triaryl- or bis(triarylamine)-functionalized bisimides show an interesting spectroelectrochemical behaviour not only in the reduction but also in the oxidation mode, associated with their quasi-reversible oxidation of their triarylamine parts to radical cations. Polymeric electrochromic devices showing high optical contrast based on this family of compounds have been proposed.24,27,28 In Fig. 8 UV-vis spectra of B2, registered for increasing working electrode potentials are shown as a representative example of the spectroelectrochemical behaviour of N-functionalized bisimides studied in this research. The remaining bisimides of this group (B1, B3, B4) behave in a similar manner, their spectra registered at different potentials can be found in Fig. S6 of ESI.† Position of the peaks characteristic of the radical cations formed are listed in Table 3. | ||
| Fig. 8 UV-vis spectra of B2 registered for increasing working electrode potential (concentration: 1 × 10−4 M; electrolyte 0.1 M Bu4NPF6 in CH2Cl2). | ||
Electrochemical oxidation of N-substituted bisimides gives rise to a profound decrease of the band at ca. 300 nm, characteristic of the neutral triarylamine moiety with simultaneous increase of two radical cation-originating bands at 568 nm and 698 nm. The bisimide band of vibronic character does not undergo significant changes, apart from slight broadening and slight increase of intensity. This means that the lowering of the electron density caused by the oxidation is limited to the triarylamine moiety, consistent with the previous DFT calculations.30,31 The observed spectral changes induced by increasing polarization of the working electrode are similar to previous results reporting spectroelectrochemical investigations of the oxidation of triarylamines linked together by a conjugated bridge.56–62
Bisimides core-substituted with triarylamine are more difficult to oxidize than the N-substituted ones (see Table 2). Consistent with the cyclic voltammetry studies, the first signs of their oxidation appear at potentials higher by ca. 50 to 90 mV as compared to the case of N-substituted bisimides. Fig. 9 shows the spectra of B5 registered at the increasing working electrode potentials. The corresponding spectra of B6 can be found in the ESI.†
 | ||
| Fig. 9 UV-vis spectra of B5 registered for increasing working electrode potential (concentration: 1 × 10−4 M; electrolyte 0.1 M Bu4NPF6 in CH2Cl2). | ||
Upon oxidation of B5 the band at 307 nm, characteristic of neutral triarylamine moiety quickly decreases with increasing electrode potential. The bisimide band at 339 nm is essentially unaffected by the oxidation, apart from an apparent decrease of its intensity caused by its partial superposition on the disappearing triarylamine band. Other changes in the UV-vis spectra are also present. The CT band at 592 nm in the experimental spectrum undergoes a hypsochromic shift to 512 nm. In addition new bands ascribed to various transitions in the radical cation form appear at a less energetic part of the spectrum. Consistent with the experimental spectrum, an oxidation induced hypsochromic shift of the CT band is also clearly seen in the calculated spectrum (see Fig. S8 in ESI†). The spectroelectrochemical behaviour of B6 is similar (see Fig. S7 in ESI†).
Spectroelectrochemical investigations in the solid state were carried out for thin films of B7. The resulting spectra are shown in Fig. 10. The observed behaviour is similar as that observed for B5 – disappearance of the triarylamine band at 313 nm and growth of a new band at 749 nm ascribed to the radical cation form of triarylamine which is superimposed on the CT band undergoing hypsochromic shift upon increasing electrode potential. The bands are however significantly broader, as frequently observed for the solid state spectra.
 | ||
| Fig. 10 UV-vis spectra of a thin film of B7 registered for increasing working electrode potential (electrolyte 0.1 M Bu4BF4/CH3CN). | ||
In all studied bisimides functionalized with triarylamine stable radical cations can be electrochemically generated. EPR spectra of the radical cation forms of B1–B4 are presented in Fig. 11a–d.
 | ||
| Fig. 11 Experimental and simulated (in the case of B4) EPR spectra of electrochemically generated radical cations of arylene bisimides N-substituted with triarylamine (1 × 10−3 M; electrolyte 0.1 M Bu4NPF6 in CH2Cl2): (a) B1 (E = 0.5 V); (b) B2 (E = 0.5 V); (c) B3 (E = 0.5 V); (d) B4 (E = 0.5 V). Modulation width in range of 0.1–0.6 G; microwave power 1 mW. Fitting parameters for B4 can be found in ESI (Chart S2†). | ||
As judged from the corresponding cyclic voltammograms in B1, B2 and B3 radical cations are formed on each triarylamine substituent at the same potential since the area under the peaks of the redox couple corresponding to the oxidation of these bisimides to the radical cation form is twice as high than the area under the peaks ascribed to the redox couple originating from the monoelectronic reduction of these bisimides to radical anions (see Fig. 1a and S1 in the ESI†). All three compounds can be considered as two triarylamine moieties connected by a non-conjugated bridge. The formation of stable dications of diradical character in this type of compounds is known,49,63–65 however the formation of such paramagnetic moieties in triarylamines connected by a conjugated linker has also been recently reported.66
Core-functionalized bisimides (B5, B6 and B7) also form stable radical cations which can be electrochemically generated and detected in an EPR spectroelectrochemical experiment. Their EPR spectra are shown in Fig. 12a–c. The spectra of radical cations of triarylamine fuctionalized arylene bisimides are less resolved as compared to the spectra of the corresponding radical anions. This is caused by the fact that in the triarylamine substituent a larger number of protons contribute to the hyperfine interaction which results in a strong overlap of individual lines and the overall line broadening. The shape of the EPR spectra is further disturbed by a somehow diradical character of the oxidized forms of B1, B2 and B3. This is clearly seen when one compares the spectra of B2 and B4 radical cations. In the case of B4 a partially resolved three-line spectrum is observed, while in B2 with two triphenylamine substituents a single broad line is seen as an effect of interactions between two unpaired spins. The obtained coupling constants for B4 seem to indicate that the maximum spin density of the unpaired electron is predominantly located on the amine nitrogen and to a lesser extent on the two alkyl-substituted rings. Smaller spin density of the unpaired electron is found on the disubstituted (phenylene) ring (ESI, Chart S2†). Thus, the unpaired electron spin density is displaced in such a manner as to be distant from the bisimide core. Poorly resolved spectra of core substituted bisimides are difficult to interpret. However, based on the value of their g-factor (see Table 5), which is similar to that found for the N-substituted bisimides, B1–B4, and to the earlier reported g-factor of the triphenylamine radical cation,67 it can be postulated that in the case of core substituted bisimides, B5–B7, the majority of the spin density is localized on the triphenylamine moiety.
 | ||
| Fig. 12 Experimental EPR spectra of electrochemically generated radical cations of arylene bisimides N-substituted with triarylamine (1 × 10−3 M; electrolyte 0.1 M Bu4NPF6 in CH2Cl2): (a) B5 (E = 0.55 V); (b) B6 (E = 0.5 V); (c) B7 (E = 0.5 V). Modulation width in range of 0.1–0.6 G; microwave power 1 mW. | ||
| Compound | g-factor |
|---|---|
| B1+ | 2.0037 |
| B2+ | 2.0037 |
| B3+ | 2.0037 |
| B4+ | 2.0038 |
| B5+ | 2.0036 |
| B6+ | 2.0038 |
| B7+ | 2.0036 |
To summarize the second part of the work, both N- and core-substituted bisimides of low molecular weight undergo quasi-reversible oxidation ascribed to the formation of stable radical cations, detectable by EPR at the triarylamine substituents. The presence of these radicals give rise to profound spectroscopic changes in the UV-vis-NIR region, distinctly different from the changes induced by electrochemical reduction of these compounds. Electrochemical oxidation of the polymeric core-functionalized bisimide is less reversible.
Conclusions
To conclude, we have studied the spectroelectrochemical behaviour of a series of arylene bisimides N- and core-substituted with triarylamine. Some of these compounds were previously investigated as components of ambipolar17,31 and p-channel30 field effect transistors. EPR spectroelectrochemical investigations confirmed ambipolarity of these compounds demonstrating the formation of stable radical anions upon quasi-reversible one-electron reduction and stable radical cations upon quasi-reversible oxidation. Both redox processes involve profound and distinctly different spectral changes in the visible range of the spectrum demonstrating their promising multicolor electrochromism.Acknowledgements
This research was supported in part by PL-Grid Infrastructure. S. Pluczyk and P. Zassowski are scholars supported by the “Doktoris–scholarship program for an innovative Silesia”, co-financed by European Union within European Social Fund. The presented research was also financed by the project entitled “New solution processable organic and hybrid (organic/inorganic) functional materials for electronics, optoelectronics and spintronics” (Contract no. TEAM/2011–8/6), which is operated within the Foundation for the Polish Science Team Programme, cofinanced by the EU European Regional Development Fund (M.Z. and A.P.) and by Polish-Norwegian Research Programme operated by the National Centre for Research and Development under the Norwegian Financial Mechanism 2009–2014 in the frame of Project Contract no. Pol-Nor/210704/43/2013 R.R). R.R. additionally acknowledges the support of the Foundation for Polish Science within the framework of START 2014 Programme.Notes and references
- Z. Chen, A. Lohr, C. R. Saha-Möller and F. Würthner, Chem. Soc. Rev., 2009, 38, 564–584 RSC.
- C. Huang, S. Barlow and S. R. Marder, J. Org. Chem., 2011, 76, 2386–2407 CrossRef CAS PubMed.
- A. N. Sokolov, M. E. Roberts, O. B. Johnson, Y. Cao and Z. Bao, Adv. Mater., 2010, 22, 2349–2353 CrossRef CAS PubMed.
- J. E. Anthony, A. Facchetti, M. Heeney, S. R. Marder and X. Zhan, Adv. Mater., 2010, 22, 3876–3892 CrossRef CAS PubMed.
- X. Zhan, A. Facchetti, S. Barlow, T. J. Marks, M. A. Ratner, M. R. Wasielewski and S. R. Marder, Adv. Mater., 2011, 23, 268–284 CrossRef CAS PubMed.
- F. Würthner and M. Stolte, Chem. Commun., 2011, 47, 5109–5115 RSC.
- R. Rybakiewicz, I. Tszydel, J. Zapala, L. Skorka, D. Wamil, D. Djurado, J. Pécaut, J. Ulanski, M. Zagorska and A. Pron, RSC Adv., 2014, 4, 14089 RSC.
- Q. Meng and W. Hu, Phys. Chem. Chem. Phys., 2012, 14, 14152–14164 RSC.
- C. R. Wade, M. Li and M. Dincă, Angew. Chem., Int. Ed. Engl., 2013, 52, 13377–13381 CrossRef CAS PubMed.
- G.-B. Li, L. Li, J.-M. Liu, T. Yang and C.-Y. Su, Cryst. Growth Des., 2013, 13, 1518–1525 CAS.
- C. F. Leong, T. B. Faust, P. Turner, P. M. Usov, C. J. Kepert, R. Babarao, A. W. Thornton and D. M. D'Alessandro, Dalton Trans., 2013, 9831–9839 RSC.
- L. Han, L. Qin, L. Xu, Y. Zhou, J. Sun and X. Zou, Chem. Commun., 2013, 49, 406–408 RSC.
- P. M. Usov, C. Fabian and D. M. D'Alessandro, Chem. Commun., 2012, 48, 3945–3947 RSC.
- R. Schmidt, J. H. Oh, Y.-S. Sun, M. Deppisch, A.-M. Krause, K. Radacki, H. Braunschweig, M. Könemann, P. Erk, Z. Bao and F. Würthner, J. Am. Chem. Soc., 2009, 131, 6215–6228 CrossRef CAS PubMed.
- H. Katz, A. Lovinger, J. Johnson, C. Kloc, T. Siegrist, W. Li, Y. Lin and A. Dodabalapur, Nature, 2000, 404, 478–481 CrossRef CAS PubMed.
- H. E. Katz, J. Johnson, A. J. Lovinger and W. Li, J. Am. Chem. Soc., 2000, 122, 7787–7792 CrossRef CAS.
- A. Pron, R. R. Reghu, R. Rybakiewicz, H. Cybulski, D. Djurado, J. V. Grazulevicius, M. Zagorska, I. Kulszewicz-Bajer and J.-M. Verilhac, J. Phys. Chem. C, 2011, 115, 15008–15017 CAS.
- Y. Ma, Y. Wu, Y. Zhao, H. Fu and J. Yao, Phys. Chem. Chem. Phys., 2011, 13, 2036–2043 RSC.
- A. Pron, P. Gawrys, M. Zagorska, D. Djurado and R. Demadrille, Chem. Soc. Rev., 2010, 39, 2577–2632 RSC.
- T. Lana-Villarreal, J. M. Campiña, N. Guijarro and R. Gómez, Phys. Chem. Chem. Phys., 2011, 13, 4013–4021 RSC.
- C. Quinton, V. Alain-Rizzo, C. Dumas-Verdes, F. Miomandre, G. Clavier and P. Audebert, RSC Adv., 2014, 4, 34332 RSC.
- P. Bujak, I. Kulszewicz-Bajer, M. Zagorska, V. Maurel, I. Wielgus and A. Pron, Chem. Soc. Rev., 2013, 42, 8895–8999 RSC.
- H.-J. Yen, C.-J. Chen and G.-S. Liou, Adv. Funct. Mater., 2013, 23, 5307–5316 CrossRef CAS.
- H.-M. Wang and S.-H. Hsiao, J. Mater. Chem. C, 2014, 2, 1553 RSC.
- H.-M. Wang and S.-H. Hsiao, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 1172–1184 CrossRef CAS.
- T. Kurosawa, Y.-C. Lai, T. Higashihara, M. Ueda, C.-L. Liu and W.-C. Chen, Macromolecules, 2012, 45, 4556–4563 CrossRef CAS.
- H.-M. Wang and S.-H. Hsiao, Polymer, 2009, 50, 1692–1699 CrossRef CAS PubMed.
- H.-J. Yen and G.-S. Liou, Polym. Chem., 2012, 3, 255 RSC.
- A. Iwan and D. Sek, Prog. Polym. Sci., 2011, 36, 1277–1325 CrossRef CAS PubMed.
- R. Rybakiewicz, D. Djurado, H. Cybulski, E. Dobrzynska, I. Kulszewicz-Bajer, D. Boudinet, J.-M. Verilhac, M. Zagorska and A. Pron, Synth. Met., 2011, 161, 1600–1610 CrossRef CAS PubMed.
- R. Rybakiewicz, J. Zapala, D. Djurado, R. Nowakowski, P. Toman, J. Pfleger, J.-M. Verilhac, M. Zagorska and A. Pron, Phys. Chem. Chem. Phys., 2013, 15, 1578–1587 RSC.
- R. Rybakiewicz, P. Gawrys, D. Tsikritzis, K. Emmanouil, S. Kennou, M. Zagorska and A. Pron, Electrochim. Acta, 2013, 96, 13–17 CrossRef CAS PubMed.
- C. M. Dufft and G. A. Heath, Inorg. Chem., 1991, 30, 2528–2535 CrossRef.
- Spectroelectrochemistry, ed. W. Kaim and A. Klein, Royal Society of Chemistry, 2008 Search PubMed.
- S. A. Macgregor, E. M. R. J. Sorbie and L. J. Yellowlees, in Molecular Electrochemistry of Inorganic, Bioinorganic and Organometallic Comounds, ed. A. J. L. Pomberio and J. A. McCleverty, 1993, pp. 503–517 Search PubMed.
- L. P. Moorcraft, A. Morandeira, J. R. Durrant, J. R. Jennings, L. M. Peter, S. Parsons, A. Turner, J. Yellowlees and N. Robertson, Dalton Trans., 2008, 6940–6947 RSC.
- D. N. Mason, G. B. Deacon, J. Yellowlees and A. M. Bond, Dalton Trans., 2003, 890–900 RSC.
- P. R. Murray, D. Collison, S. Daff, N. Austin, R. Edge, B. W. Flynn, L. Jack, F. Leroux, E. J. L. Mcinnes, A. F. Murray, D. Sells, T. Stevenson, J. Wolowska and L. J. Yellowlees, J. Magn. Reson., 2011, 213, 206–209 CrossRef CAS PubMed.
- A. Zykwinska, W. Domagala, A. Czardybon, B. Pilawa and M. Lapkowski, Electrochim. Acta, 2006, 51, 2135–2144 CrossRef CAS PubMed.
- D. R. Duling, J. Magn. Reson., Ser. B, 1994, 104, 105–110 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS PubMed.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3093 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- A.-R. Allouche, J. Comput. Chem., 2011, 32, 174–182 CrossRef CAS PubMed.
- S. K. Lee, Y. Zu, A. Herrmann, Y. Geerts, K. Müllen and A. J. Bard, J. Am. Chem. Soc., 1999, 121, 3513–3520 CrossRef CAS.
- H. Wu, H. Wang, L. Xue, Y. Shi and X. Li, J. Phys. Chem. B, 2010, 114, 14420–14425 CrossRef CAS PubMed.
- Z. An, S. A. Odom, R. F. Kelley, C. Huang, X. Zhang, S. Barlow, L. A. Padilha, J. Fu, S. Webster, D. J. Hagan, E. W. Van Stryland, M. R. Wasielewski and S. R. Marder, J. Phys. Chem. A, 2009, 113, 5585–5593 CrossRef CAS PubMed.
- M. Velusamy, J.-Y. Shen, J. T. Lin, Y.-C. Lin, C.-C. Hsieh, C.-H. Lai, C.-W. Lai, M.-L. Ho, Y.-C. Chen, P.-T. Chou and J.-K. Hsiao, Adv. Funct. Mater., 2009, 19, 2388–2397 CrossRef CAS.
- C.-C. Lin, M. Velusamy, H.-H. Chou, J. T. Lin and P.-T. Chou, Tetrahedron, 2010, 66, 8629–8634 CrossRef CAS PubMed.
- C.-C. Chao, M. Leung, Y. O. Su, K.-Y. Chiu, T.-H. Lin, S.-J. Shieh and S.-C. Lin, J. Org. Chem., 2005, 70, 4323–4331 CrossRef CAS PubMed.
- G. Andric, J. F. Boas, A. M. Bond, G. D. Fallon, K. P. Ghiggino, C. F. Hogan, J. A. Hutchison, M. A.-P. Lee, S. J. Langford, J. R. Pilbrow, G. J. Troup and C. P. Woodward, Aust. J. Chem., 2004, 57, 1011–1019 CrossRef CAS.
- M. J. Ahrens, M. J. Tauber and M. R. Wasielewski, J. Org. Chem., 2006, 71, 2107–2114 CrossRef CAS PubMed.
- A.-J. Avestro, D. M. Gardner, N. A. Vermeulen, E. A. Wilson, S. T. Schneebeli, A. C. Whalley, M. E. Belowich, R. Carmieli, M. R. Wasielewski and J. F. Stoddart, Angew. Chem., Int. Ed. Engl., 2014, 53, 4442–4449 CrossRef CAS PubMed.
- S. Barlow, C. Risko, S. Chung, N. M. Tucker, V. Coropceanu, S. C. Jones, Z. Levi, J. Bre and S. R. Marder, J. Am. Chem. Soc., 2005, 127, 16900–16911 CrossRef CAS PubMed.
- P. J. Low, M. A. J. Paterson, H. Puschmann, A. E. Goeta, A. K. Howard, C. Lambert, J. C. Cherryman, D. R. Tackley, S. Leeming and B. Brown, Chem.–Eur. J., 2004, 10, 83–91 CrossRef CAS PubMed.
- C. Lambert and S. Amthor, J. Phys. Chem. A, 2004, 108, 6474–6486 CrossRef CAS.
- C. Lambert and G. Nöll, J. Am. Chem. Soc., 1999, 121, 8434–8442 CrossRef CAS.
- J. M. Chem, C. Huang, M. M. Sartin, N. Siegel, M. Cozzuol, Y. Zhang, J. M. Hales, S. Barlow, J. W. Perry and S. R. Marder, J. Mater. Chem., 2011, 21, 16119–16128 RSC.
- M. Oyama, K. Nozaki and S. Okazaki, Anal. Chem., 1991, 63, 1387–1392 CrossRef CAS.
- C. Lambert, G. Nöll and J. Schelter, Nat. Mater., 2002, 1, 69 CrossRef CAS PubMed.
- A. Ito, M. Urabe and K. Tanaka, Angew. Chem., Int. Ed., 2009, 48, 5773–5785 CrossRef.
- Y. Yokoyama, D. Sakamaki, A. Ito, K. Tanaka and M. Shiro, Angew. Chem., Int. Ed. Engl., 2012, 51, 9403–9406 CrossRef CAS PubMed.
- C. L. Ramírez, C. Pegoraro, L. Trupp, A. Bruttomesso, V. Amorebieta, D. M. A. Vera and A. R. Parise, Phys. Chem. Chem. Phys., 2011, 13, 20076–20080 RSC.
- Y. Su, X. Wang, X. Zheng, Z. Zhang, Y. Song, Y. Sui, Y. Li and X. Wang, Angew. Chem., 2014, 126, 2901–2905 CrossRef.
- T. Nagai, K. Katayama and N. Tokura, Chem. Lett., 1973, 919–922 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra12603c |
| This journal is © The Royal Society of Chemistry 2015 |