
Metal/metal oxide nanostructures derived from metal–organic frameworks
Abstract
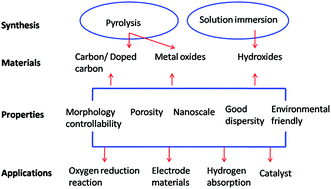
Metal–organic frameworks (MOFs) have important potential applications in gas separation, storage and purification, and also for use as electrode materials, catalysts, sensors and in drug-delivery systems. There has been increasing interest in the synthesis of micro- and nanostructures based on MOFs, particularly on the improvement of their versatility and the simplification of synthesis procedures. This paper reviews the use of MOFs as matrices for solid-state decomposition and in the synthesis of metal/metal oxide micro- and nanostructures, porous carbon and composite materials.
 Please wait while we load your content...
Please wait while we load your content...

