
A novel three-dimensional gold catalyst prepared by simple pulse electrodeposition and its high electrochemical performance for hydrogen peroxide reduction
Ke Ye,
Dongming Zhang,
Xin Wang,
Kui Cheng and
Dianxue Cao*
Key Laboratory of Superlight Materials and Surface Technology of Ministry of Education, College of Materials Science and Chemical Engineering, Harbin Engineering University, Harbin, 150001, P.R. China. E-mail: caodianxue@hrbeu.edu.cn; Fax: +86-451-82589036; Tel: +86-451-82589036
First published on 4th December 2014
Abstract
Novel Au nanoparticles (NP), Au pinecones (PC) and Au nanodendrites (ND) supported on carbon coated titanium dioxide (C@TiO2) nanoarrays were successfully obtained through a facile chemical vapor deposition of three-dimensional (3D) C@TiO2 substrate, followed by potential pulse electrodeposition of Au electrocatalysts. The morphology and structure of the open 3D Au–C@TiO2 electrodes was characterized by scanning electron microscopy and X-ray diffractometry. The different morphology of electrodeposited Au can be easily controlled by the applied potential (Eo). Electrochemical methods, including cyclic voltammetry, linear sweep voltammetry and chronoamperometry, were used to examine the catalytic activity of the electrode for H2O2 electroreduction in H2SO4 solution. The Au ND–C@TiO2 electrode exhibited the largest effective specific surface area among the Au–C@TiO2 electrodes, owing to its open nanodendritic structure allowing the full utilization of Au surface active sites. A nearly constant reduction current density of 0.655 A cm−2 was successfully achieved on the Au ND–C@TiO2 electrode at the potential of 0 V in 2.0 mol L−1 H2O2 + 2.0 mol L−1 H2SO4 solution, which was significantly higher than the catalytic activity of H2O2 electroreduction achieved previously with precious metals as catalysts.
1. Introduction
Nowadays, the demand for electrical energy has greatly increased with the rapid development of modern society and massive energy needs as well as the depletion of fossil fuels. Fuel cells (FCs) have attracted considerable attention in recent years because they can provide a promising alternative to incumbent electrical power generation technologies, for medium-scale applications such as remote or backup power, as well as small-scale applications.1–6 Over the years, the investigation of liquid-based FCs employing hydrogen peroxide (H2O2) to replace oxygen (O2) as oxidant (e.g. metal semi-FCs, direct peroxide–peroxide FCs, direct borohydride–hydrogen peroxide FCs and direct methanol–hydrogen peroxide FCs) has been a hot topic.7–28 Compared with O2, H2O2 has many advantages such as faster reduction kinetics, easier storage and feeding, the device compact and no production with toxicity during the reaction process.7–28In addition, fuel cells using H2O2 as oxidant in acid solution could achieve higher cell performance than that in alkaline medium.24–28 For example, the theoretical open circuit voltage (OCP) of direct borohydride FCs (DBFCs) with the electroreduction of H2O2 in acid is as high as 3.016 V, which is 0.9 V higher than that in basic medium. Walsh et al.24 exploited a basic-acidic bipolar electrolytes DBFC configuration, with NaBH4 electrooxidation in NaOH and H2O2 electroreduction in HCl. A high cell performance with the OCP of 1.9 V and peak power density of 34 mW cm−2 was successfully obtained. Direct peroxide–peroxide FCs also use H2O2 as oxidant in acid solution to get a high cell performance. Sanli and Aytaç28 reported a two-compartment H2O2 fuel cell having the conventional fuel cell configuration. By separating the anode and cathode compartments and operating with basic H2O2 as fuel and acidic H2O2 as oxidant, the cell demonstrated an open circuit voltage of around 0.9 V and a peak power density of 3.8 mW cm−2. Thus, in order to further improve the cell performance using H2O2 as oxidant, the electrocatalysts with high performance for H2O2 electroreduction in acid are imperious demands.
At present, macrocycle complexes of transition metals (e.g. Fe– and Co–porphyrin, Cu–triazine complexes13,14) and transition metal oxides (e.g. cobalt oxides, ferric oxides15,16) are widely used for H2O2 electroreduction due to their low cost. However, they suffer the drawback of inferior catalytic activity and unstability in acid medium. Noble metals (e.g. platinum, palladium, iridium, gold, silver and their alloys8,10–12,17–23) are the most effective catalysts for the electroreduction of H2O2 due to their high activity and superior stability in harsh acid and alkaline solution, but their applied range is greatly limited by their high price. So improving the specific surface area and then reducing the use level of noble metals are very interesting.
Generally, noble metals are loaded on carbon black to form powder catalysts. They are mixed with conducting carbons and polymer binders to form pastes, and then applied to a carbon paper current collector. Such obtained electrodes usually suffer drawbacks of low catalyst utilization because some catalysts are unable to contact with the current collector and electrolyte to form the three-phase reaction zone.29–31 Furthermore, if gas products were involved (e.g. methanol oxidation, borohydride hydrolysis), they may block the active sites of catalysts causing a reduction of catalytic efficiency due to the slow removal of gas bubbles from compact electrodes. The existence of binder will also greatly decrease the electrical conductivity of the electrode materials, reducing their electrochemical performance. To overcome these problems, one possible solution is preparing the three dimensional (3D) structured electrocatalysts directly supported on an open structural current-collecting substrate without any conductive agent and binder.
Recently, transition metal oxide nanoarrays (e.g. TiO2, α-Fe2O3, ZnO, Co3O4, Co3O4@NiO, CuO/ZnO nanoarrays)15,32–37 directly grown on the substrate have been in various applications, such as catalysts, electrochemical capacitors, lithium ion batteries and solar cells, because nanoarrays with the open structure usually possess a larger electrochemical active surface area, a higher utilization efficiency of the active materials, and a superior mass transport property. However, pure transition metal oxide has the disadvantages of poor electrical conductivity and instability in acid media.38,39 As a consequence, transition metal oxide nanoarrays coated by highly conductive carbon are urgently required as an excellent current collector.
In this work, nanoarrays consisting of carbon coated titanium dioxide (C@TiO2) by a one-step chemical vapor deposition without any template were used as the high conductive skeleton for Au deposits using potential pulse electrodeposition. Au was selected as the catalyst because it is a non-platinum catalyst with good stability in the harsh acidic H2O2 solution. Moreover, the Ti foil substrate and C@TiO2 nanoarrays are also stable in acid. C@TiO2 nanoarrays with open structures were used both as the support and the current collector to enable the electrode to have good mass transport property. The electrodes with different structural Au catalysts (nanoparticles, pinecones and nanodendrites) were obtained by varying the electrodeposition potentials. The Au loading and the electrodes for the electrochemical performance of H2O2 reduction were systematically investigated. The Au nanodendrites deposited on the surface of C@TiO2 (Au ND–C@TiO2) electrode demonstrated the highest activity, Au utilization and catalytic performance for H2O2 electroreduction among the three electrodes in H2SO4 solution.
2. Experimental
2.1. Reagents
Chloroauric acid tetrahydrate (HAuCl4·4H2O) (>99.9%), sulphuric acid (H2SO4), hydrogen peroxide (H2O2), acetone (CH3COCH3), isopropanol ((CH3)2CHOH), ethanol (C2H5OH), hydrofluoric acid (HF) and nitric acid (HNO3) were obtained from Enterprise Group Chemicals Reagent Co. Ltd China. Ti foil (thickness: 1 mm) was purchased from Baoji Yiyuan titanium industry Co., Ltd All chemicals are analytical grade and were used as-received without further purification. Ultra-pure water (Millipore, 18 MΩ cm) was used throughout the study.2.2. Preparation and characterization of Au–C@TiO2 electrodes
The schematic illustration for the synthesis of Au–C@TiO2 electrodes is shown in Fig. 1a. The C@TiO2 nanoarrays were synthesized according to the procedure reported by Huo et al.39 In brief, Ti foil (10 mm × 10 mm × 1 mm) was degreased ultrasonically in acetone, isopropanol and ethanol sequentially, followed by polishing in a solution containing H2O, HF and HNO3 with a volume ratio of 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:4 for 5 min to remove the surface native oxide layer. After rinsing with deionized water and drying under flowing N2 (99.999%) gas, the Ti foil was loaded onto a ceramic substrate placed in the center of an alumina tube inside a horizontal tube furnace. The reactor was purged with argon several times to remove residual oxygen and/or moisture before being heated to 850 °C under Ar (99.9999%) gas. Acetone was subsequently introduced into the chamber by argon at a flow rate of 150 SCCM (SCCM denotes standard cubic centimeter per minute at STP). The reaction proceeded for 1.5 h and then the sample was cooled to room temperature under argon gas.
:1:4 for 5 min to remove the surface native oxide layer. After rinsing with deionized water and drying under flowing N2 (99.999%) gas, the Ti foil was loaded onto a ceramic substrate placed in the center of an alumina tube inside a horizontal tube furnace. The reactor was purged with argon several times to remove residual oxygen and/or moisture before being heated to 850 °C under Ar (99.9999%) gas. Acetone was subsequently introduced into the chamber by argon at a flow rate of 150 SCCM (SCCM denotes standard cubic centimeter per minute at STP). The reaction proceeded for 1.5 h and then the sample was cooled to room temperature under argon gas.
 | ||
| Fig. 1 Schematic diagram of the preparation process of Au–C@TiO2 electrodes (a) and the potential pulse electrodeposition (b). | ||
The Au NP–C@TiO2, Au PC–C@TiO2 and Au ND–C@TiO2 electrodes were fabricated by potential pulse electrodeposition of Au nanoparticles, pinecones and nanodendrites directly on the C@TiO2 nanoarrays (Fig. 1a). The electrodeposition was carried out in a standard three-electrode electrochemical cell controlled by computerized potentiostat (Autolab PGSTAT302, Eco Chemie). A piece of C@TiO2 substrate was employed as the working electrode, and a platinum foil (10 × 10 mm) was served as the counter electrode. A saturated Ag/AgCl (3 mol L−1 KCl) electrode was used as the reference electrode, and all potentials in this work were referred to this reference electrode. The solution contains 0.5 mmol L−1 HAuCl4 and 0.5 mol L−1 H2SO4. Before electrodeposition, a piece of C@TiO2 substrate was first immersed in HNO3 solution for 30 min, and then the Au nanoparticles, pinecones and nanodendrites were achieved by potential pulse electrodeposition method (Fig. 1b). The reduction potential (Er) was fixed at −0.5 V, and the oxidation potential (Eo) was changed to be 0.7, 0.9 and 1.1 V, respectively. The frequency was 5 Hz and the deposition time was 30 min. The electrolyte was kept stirring under flowing nitrogen during the electrodeposition. After deposition, the electrode was removed from the solution and washed with ultrapure water thoroughly, and air-dried prior.
The Au–C@TiO2 electrodes were characterized by a scanning electron microscope (SEM, JEOL JSM-6480). The structure was analyzed using an X-ray diffractometer (Rigaku TTR III) with Cu-Kα radiation (λ = 0.1514178 nm). The Au loading was measured using an inductive coupled plasma emission spectrometer (ICP, Xseries II, Thermo Scientific). Au in the 1.0 cm2 electrode was first dissolved in aqua regia solution and then diluted to 1 L solution for the ICP measurement.
2.3. Electrochemical measurements
The electrochemical measurements were also performed in a standard three-electrode electrochemical cell with saturated Ag/AgCl (3 mol L−1 KCl) reference electrode and Pt counter electrode. The electrolyte for H2O2 electroreduction was H2O2 containing H2SO4. The electrolyte solutions were deoxygenated by bubbling ultrahigh purity N2 for 15 min and maintained under N2 atmosphere during measurements. All measurements were performed at ambient temperature (20 ± 2 °C). Linear sweep voltammetry (LSV), cyclic voltammetry (CV) and chronoamperometry (CA) were conducted using a computerized potentiostat (Autolab PGSTAT302, Eco Chemie) controlled by GPES software. The reported current densities were calculated using the geometrical area of the electrode.3. Results and discussion
3.1. Characterization of Au–C@TiO2 electrodes
The Au–C@TiO2 electrodes were prepared by potential pulse electrodeposition of Au electrocatalysts directly on the C@TiO2 nanoarrays at different applied potentials (Eo). Fig. 2 shows the SEM images with different magnification of Au NP–C@TiO2, Au PC–C@TiO2, Au ND–C@TiO2 electrodes obtained at Eo of 0.7 V, 0.9 V and 1.1 V, respectively. The C@TiO2 nanoarrays were also shown for comparison. Ti foil as the substrate for the direct growth of C@TiO2 nanoarrays can be achieved many advantages including high stability in the harsh acid medium and easy preparation of membrane electrolyte assembly due to the ultrathin properties (the thickness of 1 mm). It can be seen from Fig. 2g that the surface of Ti foil was completely covered by the dense growth of C@TiO2 nanoarrays. The C@TiO2 nanoarrays grew almost vertically from the substrate with the length of ∼5 μm and the diameter of ∼150 nm, which provided an open three-dimensional skeleton for the electrodeposition of Au, allowing electrolytes to access the full electrode surface. Besides, the high electrical conductivity and stability of C@TiO2 nanoarrays make it a desirable support of Au catalysts and the current collector of the electrodes. Fig. 2a and b present the different magnification SEM images of typical Au NP–C@TiO2 electrode and demonstrate that a series of Au nanoparticles with the size of ∼160 nm uniformly covered the surface of C@TiO2 nanoarrays. As can be observed from Fig. 2c and d, the homogeneous and compact Au pinecones were equably formed on the surface of C@TiO2 nanoarrays after Au electrodeposition. The lower magnification SEM image of the Au ND–C@TiO2 electrode (Fig. 2e) manifested that dendritic Au was deposited on the C@TiO2 and aligned perpendicularly to the nanoarray surface. The width of these dendritic nanostructures was hundreds of nanometers and the length was up to about 0.8 μm. There were also some particle-like structures on the surface of C@TiO2 nanoarrays in the high magnification SEM image (Fig. 2f). Higher magnification SEM image (insert of Fig. 2f) revealed the detail of an Au dendrite. The single Au dendrite was composed of a backbone with symmetrical side branches. The diameters of both the trunks and side branches were less than 100 nm. This unique structure of the Au ND–C@TiO2 electrode ensured the full utilization of Au surfaces because all of the Au surfaces are accessible to electrolytes. The results show that the surface morphology of Au electrocatalysts changes from nanoparticles prepared at Eo of 0.7 V to pinecone-like morphology obtained at Eo of 0.9 V and finally to dendritic morphology deposited at Eo of 1.1 V with the increase of Eo. Consequently, under the condition of potential pulse electrodeposition, the morphologies of Au electrocatalysts can be adjusted simply by changing the applied oxidation potential. Above all, the obtained Au NP, Au PC and Au ND–C@TiO2 electrodes may be particularly suitable for H2O2 electroreduction in acid medium, making electroactive Au with a large bare surface place in direct contact with the electrolyte. | ||
| Fig. 2 Low-magnification and high-magnification SEM images of Au NP–C@TiO2 electrode (a and b), Au PC–C@TiO2 electrode (c and d), Au ND–C@TiO2 electrode (e and f) and C@TiO2 nanoarrays (g). | ||
Based on the results obtained by scanning electron microscopy, X-ray diffraction analysis was employed to further identify the structure of the Au–C@TiO2 electrodes. Fig. 3 displays the XRD patterns of Au NP–C@TiO2, Au PC–C@TiO2 and Au ND–C@TiO2 electrode, along with that of the bare C@TiO2 nanoarrays for comparison. The pattern of Au ND–C@TiO2 electrode exhibited four diffraction peaks at 2θ = 38.2°, 44.4°, 64.5° and 77.5°, which can be indexed to the diffraction from the (111), (200), (220) and (311) plane of the face-centered cubic Au metal, respectively, according to the standard crystallographic spectrum of Au (JCPDS card no. 65-2870), indicating that Au nanodendrites present as the metallic state. XRD patterns of the Au PC–C@TiO2 and Au NP–C@TiO2 electrode also displayed the characteristic peak of metallic Au. Differing from the Au ND–C@TiO2 electrode, the peak intensities of (111), (200), (220) and (311) plane of Au–C@TiO2 electrodes obviously decreased in the order of Au ND–C@TiO2, Au PC–C@TiO2 and Au NP–C@TiO2 electrode, suggesting that the Au nanodendrites include the best well-crystallized gold nanocrystals among the three electrodes. Furthermore, the intensities of the main diffraction peaks located at 2θ = 27.3° and 39.8° for C@TiO2 substrate were dramatically diminished after being covered by Au nanoparticles, pinecones and nanodendrites, revealing that Au nanoparticles, pinecones and nanodendrites were uniformly deposited upon the C@TiO2.
 | ||
| Fig. 3 XRD patterns of the C@TiO2 nanoarrays (a), Au NP–C@TiO2 electrode (b), Au PC–C@TiO2 electrode (c) and Au ND–C@TiO2 electrode (d). | ||
3.2. Electrocatalytic performance of H2O2 electroreduction on the Au–C@TiO2 electrodes
Fig. 4 exhibits the typical cyclic voltammograms (CVs) of the C@TiO2, Au NP–C@TiO2, Au PC–C@TiO2, Au ND–C@TiO2 electrodes measured in 1.0 mol L−1 H2SO4 at a scan rate of 50 mV s−1. It is clear that no obvious oxidation/reduction peaks were observed on the C@TiO2 substrate, indicating that C@TiO2 was stable in acid. Differing from the CV of C@TiO2 substrate, the hydrogen adsorption/desorption peaks and surface oxide formation/reduction peaks are readily seen in the CVs of Au NP, Au PC and Au ND–C@TiO2 electrodes. It displays a typical response of polycrystalline Au in H2SO4 solution. The anodic peaks starting from 1.04 V is due to the oxidation of surface Au leading to the formation of an Au surface oxide layer. The cathodic peak centered at 0.98 V corresponds to the reduction of Au surface oxides.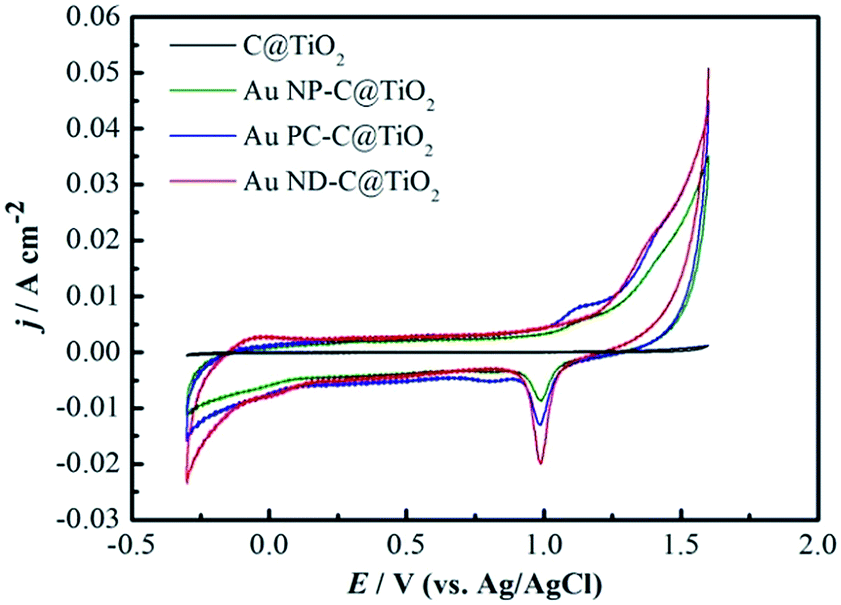 | ||
| Fig. 4 Cyclic voltammograms of the C@TiO2 substrate, Au NP–C@TiO2 electrode, Au PC–C@TiO2 electrode and Au ND–C@TiO2 electrode in 1.0 mol L−1 H2SO4 at a scan rate of 50 mV s−1. | ||
Assuming a monolayer of AuO was formed on Au surface in the positive potential scan, the electrochemically active surface area (EASA) of Au catalyst can be estimated from the charge corresponding to the cathodic reduction peak of surface oxide.40,41 Based on the monolayer charge of 390 μC cm−2 for AuO, it can be calculated that the EASA of Au NP–C@TiO2, Au PC–C@TiO2 and Au ND–C@TiO2 electrode per cm2 (geometrical area) was 53.3, 76.6 and 91.7 cm2, respectively. The EASAs of Au ND and Au PC–C@TiO2 electrode were ∼1.7 times and 1.4 times larger than that of Au NP–C@TiO2 electrode, respectively. By contrast, the value of both Au ND and Au PC–C@TiO2 electrodes mentioned here was much larger than that of dendritic Au and Pd prepared in our previous work.23,42 ICP measurement shows that the loading of Au in the Au NP, Au PC and Au ND–C@TiO2 electrode was 0.1342, 0.1486 and 0.1507 mg cm−2, respectively. So the effective specific surface area (ESSA) of Au reached 39.7, 51.5 and 60.8 m2 g−1 for Au nanoparticles, Au pinecones and Au nanodendrites, respectively. Therefore, the effective specific surface area of Au was increased by forming pinecones and nanodendrites. In addition, the hydrogen adsorption/desorption current on the Au ND–C@TiO2 electrode could be more clearly observed than that on the Au NP–C@TiO2 and Au PC–C@TiO2 at −0.2 V. This implies that nanodendritic Au has higher electrocatalytic activity than Au pinecones and Au nanoparticles. Moreover, the ESSA value of nanodendritic Au on the C@TiO2 was much larger than that of nano-Au and nano-Pd supported on carbon materials (carbon fiber cloth, carbon black, carbon nanotubes) reported in the literatures.23,42–44 The large surface area of Au ND–C@TiO2 electrode was most possibly provided by the excellent skeleton of C@TiO2 nanoarrays and its dendritic structure. Importantly, high ESSA usually means high utilization of Au, which is very important for the reduction of the cost of precious metal catalysts.
In order to further confirm that the Au ND–C@TiO2 electrode is the superior electrocatalyst to Au NP and Au PC–C@TiO2 electrodes, the comparative polarization curves for H2O2 electroreduction in H2SO4 on the different electrodes were investigated in Fig. 5a. The linear sweep voltammograms (LSVs) of C@TiO2 substrate in 1.0 mol L−1 H2SO4 and 1.0 mol L−1 H2O2 was also shown for comparison. The C@TiO2 substrate exhibited no catalytic activity for H2O2 electroreduction, revealing that the high performance was contributed to the Au deposits. The current densities at the same potential increased significantly in the sequence of Au NP, Au PC and Au ND–C@TiO2, indicating that the Au ND–C@TiO2 electrode had higher electrocatalytic performance than the Au NP and Au PC–C@TiO2 electrodes for H2O2 electroreduction. This result is consistent with the order of ESSA (Au nanodendrites > Au pinecones > Au nanoparticles).
 | ||
| Fig. 5 (a) Comparative polarization curves for H2O2 electroreduction on the C@TiO2 substrate, Au NP–C@TiO2, Au PC–C@TiO2 and Au ND–C@TiO2 electrode in 1.0 mol L−1 H2SO4 + 1.0 mol L−1 H2O2 solution at a scan rate of 5 mV s−1; (b) chronoamperometric curves for H2O2 electroreduction on the Au NP–C@TiO2, Au PC–C@TiO2 and Au ND–C@TiO2 electrode in 1.0 mol L−1 H2SO4 + 1.0 mol L−1 H2O2 solution at the potential of 0 V. | ||
Chronoamperometry was carried out to further study the optimal Au–C@TiO2 electrode for catalyzing H2O2 electroreduction. Fig. 5b shows the chronoamperometric curves (CAs) for H2O2 electroreduction in H2SO4 on the Au–C@TiO2 electrodes. As can be seen, the reduction current density of all the Au–C@TiO2 electrodes reached to steady state after a few seconds and displayed no sign of decrease within 1000 s test period at the potential of 0 V, indicating that all the electrodes have a superior stability for H2O2 electroreduction in acid medium. In addition, no obvious chemical decomposition of H2O2 was observed during the tests. It is clear that the current density on the Au ND–C@TiO2 electrode kept at 0.347 A cm−2, but that on the Au NP and Au PC–C@TiO2 electrode was just 0.202 and 0.283 A cm−2. The current densities of Au ND and Au PC–C@TiO2 electrode were ∼1.7 times and 1.4 times higher than that of Au NP–C@TiO2 electrode, respectively. The ratios of the current densities were proportional to the corresponding ratios of the EASAs, indicating that the specific catalytic activity might be the same for the three electrocatalysts. It also demonstrated that the optimum Au–C@TiO2 electrode was Au nanodendrites and the result was in good accordance with the LSV test (Fig. 5a).
3.3. Effects of H2SO4 and H2O2 concentration for H2O2 electrochemical reduction on the Au ND–C@TiO2 electrode
Karl J. J. Mayrhofer et al.45–49 found that the electrolyte anions and concentration had a large impact on the electrochemical reduction of hydrogen peroxide, and the electroactivity of the anions decreased in the order ClO4− > HSO4− > Cl− > Br− > I−. Considering the instability of ClO4−, H2SO4 was selected as the electrolyte in this work. In order to identify an appropriate H2SO4 concentration for Au ND–C@TiO2 electrode, LSV and CA measurements were examined. The concentration of H2SO4 was changed with H2O2 concentration constantly kept at 2.0 mol L−1. Fig. 6 presents the influence of H2SO4 concentration for H2O2 electroreduction on the Au ND–C@TiO2 electrode. It can be observed from Fig. 6a that the onset reduction potentials were ∼0.58 V and the reduction current density similarly increased with the negative scan going in the potential range of 0.6 to −0.2 V. Furthermore, excess electrolyte made no contribution to the enhancement of the activity of H2O2 reduction. H2SO4 concentration of 2.0 mol L−1 yielded the best performance. CAs for H2O2 electroreduction were also studied in detail (Fig. 6b). At the potential of 0 V, a nearly constant reduction current density of all the different concentration H2SO4 was achieved on the Au ND–C@TiO2 electrode within 1000 s test period, suggesting that the electrode has a good stability for H2O2 electroreduction. The result revealed that the current density with 2.0 mol L−1 H2SO4 kept at 0.655 A cm−2, while the current density with 1.0 and 3.0 mol L−1 H2SO4 was 0.428 and 0.537 A cm−2. It was also displayed that the best suitable H2SO4 concentration was 2.0 mol L−1. | ||
| Fig. 6 (a) Linear sweep voltammograms for H2O2 electroreduction on the Au ND–C@TiO2 electrode in x mol L−1 H2SO4 + 2.0 mol L−1 H2O2 (x = 1.0, 2.0 and 3.0, scan rate: 5 mV s−1); (b) chronoamperometric curves for H2O2 electroreduction at the potential of 0 V in 2.0 mol L−1 H2O2 with different H2SO4 concentrations. | ||
Fig. 7a shows the dependence of H2O2 concentration for H2O2 reduction on the Au ND–C@TiO2 electrode. The current density increased dramatically with increasing the H2O2 concentration from 0.5 to 1.5 mol L−1 at the same potential, but the further increase of H2O2 concentration from 1.5 to 2.0 mol L−1 only leads to a slight increase of current density. It is worth to mention here that the chemical decomposition of H2O2 can be discovered as the H2O2 concentration was higher than 2.0 mol L−1, which not only causes the waste of oxidant but also the bubbles produced from H2O2 decomposition can block the mass transport and active sites of catalyst. The Au ND–C@TiO2 electrode showed a reduction current density of 369 mA cm−2 at 0 V in the solution of 2.0 mol L−1 H2SO4 + 0.5 mol L−1 H2O2, which was much higher than the commercial Pd/C electrode with a Pd loading of 0.31 mg cm−2 and other precious metal composite electrodes reported in the literatures.22,23,50,51 The high electrocatalytic performance of the commercial Pd/C, Au or Pd composite electrodes and Au ND–C@TiO2 electrode was also summarized and compared in the Table 1. By comparison with the commercial Pd/C, Pd/CFC, Au–Pd NPs/CFC, Au@Pd/CFC, Au/CFC, Pd nanofilm electrodes previously reported, nanodendritic Au on the C@TiO2 nanoarrays exhibited significantly higher catalytic property to H2O2 electroreduction in acid, which is owing to the unique open 3D skeleton of C@TiO2 nanoarrays, enabling the full utilization of Au surfaces and making the electrode have higher electrochemical activity.
 | ||
| Fig. 7 (a) Linear sweep voltammograms for H2O2 electroreduction on the Au ND–C@TiO2 electrode in 2.0 mol L−1 H2SO4 + x mol L−1 H2O2 (x = 0.5, 1.0, 1.5 and 2.0, scan rate: 5 mV s−1); (b) chronoamperometric curves for H2O2 electroreduction at the potential of 0 V in 2.0 mol L−1 H2SO4 with different H2O2 concentrations. | ||
| H2O2 concentration (mol L−1) | 0.5 | 1.0 | 1.5 | 2.0 | Ref. |
|---|---|---|---|---|---|
| Commercial Pd/C electrode | 95 | — | — | 112 | 22 and 23 |
| Pd/CFC electrode | 152 | 294 | 398 | 480 | 23 |
| Au–Pd NPs/CFC electrode | 183 | 326 | 357 | 388 | 50 |
| Au@Pd/CFC electrode | 205 | — | — | — | 51 |
| Au/CFC | 246 | — | — | — | 42 |
| Pd nanofilm electrode | 213 | 329 | 404 | 415 | 22 |
| Au ND–C@TiO2 electrode | 369 | 468 | 590 | 655 | This work |
Fig. 7b exhibits the CAs with different H2O2 concentrations at the potential of 0 V. The reduction current density reached to the steady state after the initial current decay at all the different H2O2 concentrations within 1000 s test period, which indicated that the Au ND–C@TiO2 electrode possesses an excellent stability for H2O2 electroreduction in acid media.
The stability of Au ND–C@TiO2 electrode for H2O2 electroreduction at different applied potential was investigated by chronoamperometric experiment. Fig. 8 shows CAs of H2O2 electroreduction at different applied potential in 2.0 mol L−1 H2SO4 + 2.0 mol L−1 H2O2. After a rapid initial wave, the current density reached steady state at the different control potentials (0.2, 0 and −0.2 V) with slightly decreasing in 1000 s test period. In addition, the current density increased with changing the potential to more negative direction, which was in good agreement with the LSV results shown in Fig. 7a. The final current densities after 1000 s reaction at 0.2, 0 and −0.2 V were 0.418, 0.637 and 0.836 A cm−2, respectively. Notably, all of the above electrochemical tests were carried out by using the same electrode and the total time in the testing solution was about 8 hours and based on the stable CAs, it can be concluded that the nanodendritic Au on the C@TiO2 has superior electrochemical stability and is promising catalytic materials for fuel cells employing H2O2 as oxidant in acid.
 | ||
| Fig. 8 Chronoamperometric curves for H2O2 electroreduction at different potentials in 2.0 mol L−1 H2SO4 + 2.0 mol L−1 H2O2. | ||
4. Conclusions
A novel three-dimensional electrode facilely fabricated by the potential pulse electrodepositing Au nanoparticles, Au pinecones and Au nanodendrites on the surface of C@TiO2 nanoarrays are employed for H2O2 electroreduction and exhibit high catalytic performance and superior stability. The surface morphologies of Au electrocatalysts on the C@TiO2 plays an important role in determining the electrocatalytic activity for H2O2 electroreduction in acid and they are strongly dependent on the applied oxidation potential (Eo) of pulsed electrodeposition. With the increase of Eo, the surface morphology of Au electrocatalysts changes from nanoparticles prepared at Eo of 0.7 V to pinecone-like morphology obtained at Eo of 0.9 V and finally to dendritic morphology deposited at Eo of 1.1 V. The dendritic Au on the C@TiO2 possesses much larger ESSA for the H2O2 electroreduction, followed by the Au with pinecone-like morphology while the smooth Au nanoparticles have the smallest ESSA. The Au ND–C@TiO2 electrode related to its special morphology and large ESSA exhibited the highest electrocatalytic activity among the Au–C@TiO2 electrodes, which has great significance to lower the use of noble metal. This unique open structure of the electrode enables the full utilization of Au surfaces and allows the easy transportation of reactants to the catalyst as well as the quick removal of gaseous products from the electrode. In summary, an original Au ND–C@TiO2 electrode can be candidate for the promising application in fuel cells employed H2O2 as oxidant in acid.Acknowledgements
We gratefully acknowledge the financial support of this research by the National Natural Science Foundation of China (21403044), the Heilongjiang Postdoctoral Fund (LBH-Z13059), the China Postdoctoral Science Foundation (2014M561332) and the Fundamental Research Funds for the Central Universities (HEUCF201403018).Notes and references
- O. Z. Sharaf and M. F. Orhan, An overview of fuel cell technology: Fundamentals and applications, Renewable Sustainable Energy Rev., 2014, 32, 810 CrossRef CAS PubMed.
- D. S. Falcão, V. B. Oliveira, C. M. Rangel and A. M. F. R. Pinto, Review on micro-direct methanol fuel cells, Renewable Sustainable Energy Rev., 2014, 34, 58 CrossRef PubMed.
- P. Pei and H. Chen, Main factors affecting the lifetime of Proton Exchange Membrane fuel cells in vehicle applications: A review, Appl. Energy, 2014, 125, 60 CrossRef PubMed.
- M. Yetano Roche, S. Mourato, M. Fischedick, K. Pietzner and P. Viebahn, Public attitudes towards and demand for hydrogen and fuel cell vehicles: A review of the evidence and methodological implications, Energy Policy, 2010, 38, 5301 CrossRef PubMed.
- S. Kakaç, A. Pramuanjaroenkij and X. Y. Zhou, A review of numerical modeling of solid oxide fuel cells, Int. J. Hydrogen Energy, 2007, 32, 761 CrossRef PubMed.
- X. Xu, P. Li and Y. Shen, Small-scale reforming of diesel and jet fuels to make hydrogen and syngas for fuel cells: A review, Appl. Energy, 2013, 108, 202 CrossRef CAS PubMed.
- Ø. Hasvold, K. H. Johansen, O. Mollestad, S. Forseth and N. Størkersen, The alkaline aluminium/hydrogen peroxide power source in the Hugin II unmanned underwater vehicle, J. Power Sources, 1999, 80, 254 CrossRef.
- W. Yang, S. Yang, W. Sun, G. Sun and Q. Xin, Nanostructured silver catalyzed nickel foam cathode for an aluminum–hydrogen peroxide fuel cell, J. Power Sources, 2006, 160, 1420 CrossRef CAS PubMed.
- G. H. Miley, N. Luo, J. Mather, R. Burton, G. Hawkins, L. Gu, E. Byrd, R. Gimlin, P. J. Shrestha, G. Benavides, J. Laystrom and D. Carroll, Direct NaBH4/H2O2 fuel cells, J. Power Sources, 2007, 165, 509 CrossRef CAS PubMed.
- R. K. Raman, S. K. Prashant and A. K. Shukla, A 28-W portable direct borohydride–hydrogen peroxide fuel-cell stack, J. Power Sources, 2006, 162, 1073 CrossRef CAS PubMed.
- D. N. Prater and J. J. Rusek, Energy density of a methanol/hydrogen–peroxide fuel cell, Appl. Energy, 2003, 74, 135 CrossRef CAS.
- F. Yang, K. Cheng, X. Liu, S. Chang, J. Yin, C. Du, L. Du, G. Wang and D. Cao, Direct peroxide–peroxide fuel cell – Part 2: Effects of conditions on the performance, J. Power Sources, 2012, 217, 569 CrossRef CAS PubMed.
- V. L. N. Dias, E. N. Fernandes, L. M. S. da Silva, E. P. Marques, J. Zhang and A. L. B. Marques, Electrochemical reduction of oxygen and hydrogen peroxide catalyzed by a surface copper(II)-2,4,6-tris(2-piridil)-1,3,5-triazine complex adsorbed on a graphite electrode, J. Power Sources, 2005, 142, 10 CrossRef CAS PubMed.
- R. K. Raman and A. K. Shukla, Electro-reduction of hydrogen peroxide on iron tetramethoxy phenyl porphyrin and lead sulfate electrodes with application in direct borohydride fuel cells, J. Appl. Electrochem., 2005, 35, 1157 CrossRef CAS.
- D. Cao, J. Chao, L. Sun and G. Wang, Catalytic behavior of Co3O4 in electroreduction of H2O2, J. Power Sources, 2008, 179, 87 CrossRef CAS PubMed.
- R. X. Feng, H. Dong, Y. D. Wang, X. P. Ai, Y. L. Cao and H. X. Yang, A simple and high efficient direct borohydride fuel cell with MnO2-catalyzed cathode, Electrochem. Commun., 2005, 7, 449 CrossRef CAS PubMed.
- L. Gu, N. Luo and G. H. Miley, Cathode electrocatalyst selection and deposition for a direct borohydride/hydrogen peroxide fuel cell, J. Power Sources, 2007, 173, 77 CrossRef CAS PubMed.
- R. R. Bessette, M. G. Medeiros, C. J. Patrissi, C. M. Deschenes and C. N. LaFratta, Development and characterization of a novel carbon fiber based cathode for semi-fuel cell applications, J. Power Sources, 2001, 96, 240 CrossRef CAS.
- M. G. Medeiros and E. G. Dow, Magnesium-solution phase catholyte seawater electrochemical system, J. Power Sources, 1999, 80, 78 CrossRef CAS.
- H. Liu, L. Zhang, J. Zhang, D. Ghosh, J. Jung, B. W. Downing and E. Whittemore, Electrocatalytic reduction of O2 and H2O2 by adsorbed cobalt tetramethoxyphenyl porphyrin and its application for fuel cell cathodes, J. Power Sources, 2006, 161, 743 CrossRef CAS PubMed.
- K. Cheng, F. Yang, Y. Xu, L. Cheng, Y. Bao, D. Cao and G. Wang, Pd doped Co3O4 nanowire array as the H2O2 electroreduction catalyst, J. Power Sources, 2013, 240, 442 CrossRef CAS PubMed.
- K. Cheng, F. Yang, D. Zhang, J. Yin, D. Cao and G. Wang, Pd nanofilm supported on C@TiO2 nanocone core/shell nanoarrays: A facile preparation of high performance electrocatalyst for H2O2 electroreduction in acid medium, Electrochim. Acta, 2013, 105, 115 CrossRef CAS PubMed.
- F. Yang, K. Cheng, Y. Mo, L. Yu, J. Yin, G. Wang and D. Cao, Direct peroxide–peroxide fuel cell – Part 1: The anode and cathode catalyst of carbon fiber cloth supported dendritic Pd, J. Power Sources, 2012, 217, 562 CrossRef CAS PubMed.
- C. P. de León, F. C. Walsh, A. Rose, J. B. Lakeman, D. J. Browning and R. W. Reeve, A direct borohydride–Acid peroxide fuel cell, J. Power Sources, 2007, 164, 441 CrossRef PubMed.
- S. A. M. Shaegh, N. T. Nguyen, S. M. M. Ehteshami and S. H. Chan, A membraneless hydrogen peroxide fuel cell using Prussian Blue as cathode material, Energy Environ. Sci., 2012, 5, 8225 Search PubMed.
- Y. Yamada, Y. Fukunishi, S. Yamazaki and S. Fukuzumi, Hydrogen peroxide as sustainable fuel: electrocatalysts for production with a solar cell and decomposition with a fuel cell, Chem. Commun., 2010, 46, 7334 RSC.
- A. E. Sanli, O. Yilmaz and A. Aytac, A novel H2S/H2O2 fuel cell operating at the temperature of 298 K, Int. J. Energy Res., 2013, 37, 1205 CrossRef CAS.
- A. E. Sanli and A. Aytac, Response to Disselkamp: Direct peroxide/peroxide fuel cell as a novel type fuel cell, Int. J. Hydrogen Energy, 2011, 36, 869 CrossRef CAS PubMed.
- N. Cheng, H. Lv, W. Wang, S. Mu, M. Pan and F. Marken, An ambient aqueous synthesis for highly dispersed and active Pd/C catalyst for formic acid electro-oxidation, J. Power Sources, 2010, 195, 7246 CrossRef CAS PubMed.
- L. Feng, S. Yao, X. Zhao, L. Yan, C. Liu and W. Xing, Electrocatalytic properties of Pd/C catalyst for formic acid electrooxidation promoted by europium oxide, J. Power Sources, 2012, 197, 38 CrossRef CAS PubMed.
- S. Ha, R. Larsen and R. I. Masel, Performance characterization of Pd/C nanocatalyst for direct formic acid fuel cells, J. Power Sources, 2005, 144, 28 CrossRef CAS PubMed.
- K. Xie, J. Li, Y. Lai, Z. Zhang, Y. Liu, G. Zhang and H. Huang, Polyaniline nanowire array encapsulated in titania nanotubes as a superior electrode for supercapacitors, Nanoscale, 2011, 3, 2202 RSC.
- K. Xie, J. Li, Y. Lai, W. Lu, Z. Zhang, Y. Liu, L. Zhou and H. Huang, Highly ordered iron oxide nanotube arrays as electrodes for electrochemical energy storage, Electrochem. Commun., 2011, 13, 657 CrossRef CAS PubMed.
- X. H. Xia, Y. S. Luo, Z. Wang, Y. Liang, J. Fan, Z. J. Jia and Z. H. Chen, Ultrasonic synthesis and photocatalytic activity investigation of TiO2 nanoarrays, Mater. Lett., 2007, 61, 2571 CrossRef CAS PubMed.
- G. X. Qin, Q. Zou, B. Dong, H. K. Ni, W. T. Liu and G. P. Tu, Pipelinedflash-synthesis of patterned ZnO nanoarrays and nanodevices, Ceram. Int., 2014, 40, 9671 CrossRef CAS PubMed.
- T. Soejima, K. Takada and S. Ito, Alkaline vapor oxidation synthesis and electrocatalytic activity toward glucose oxidation of CuO/ZnO composite nanoarrays, Appl. Surf. Sci., 2013, 277, 192 CrossRef CAS PubMed.
- Q. Yang, Z. Lu, T. Li, X. Sun and J. Liu, Hierarchical construction of core–shell metal oxide nanoarrays with ultrahigh areal capacitance, Nano Energy, 2014, 7, 170 CrossRef CAS PubMed.
- K. Xie, Z. Lu, H. Huang, W. Lu, Y. Lai, J. Li, L. Zhou and Y. Liu, Iron supported C@Fe3O4 nanotube array: a new type of 3D anode with low-cost for high performance lithium-ion batteries, J. Mater. Chem., 2012, 22, 5560 RSC.
- K. Huo, X. Zhang, L. Hu, X. Sun, J. Fu and P. K. Chu, One-step growth and field emission properties of quasialigned TiO2 nanowire/carbon nanocone core–shell nanostructure arrays on Ti substrates, Appl. Phys. Lett., 2008, 93, 013105 CrossRef PubMed.
- Y. Liu, Y. Zeng, R. Liu, H. Wu, G. Wang and D. Cao, Poisoning of acetone to Pt and Au electrodes for electrooxidation of 2-propanol in alkaline medium, Electrochim. Acta, 2012, 76, 174 CrossRef CAS PubMed.
- R. F. Carvalhal, R. S. Freire and L. T. Kubota, Polycrystalline gold electrodes: a comparative study of pretreatment procedures used for cleaning and thiol self-assembly monolayer formation, Electroanalysis, 2005, 17, 1251 CrossRef CAS.
- F. Yang, K. Cheng, T. Wu, Y. Zhang, J. Yin, G. Wang and D. Cao, Preparation of Au nanodendrites supported on carbon fiber cloth and its catalytic performance to H2O2 electroreduction and electrooxidation, RSC Adv., 2013, 3, 5483 RSC.
- R. Rego, C. Oliveira, A. Velázquez and P. L. Cabot, A new route to prepare carbon paper-supported Pd catalyst for oxygen reduction reaction, Electrochem. Commun., 2010, 12, 745 CrossRef CAS PubMed.
- C. Hu, Z. Bai, L. Yang, J. Lv, K. Wang, Y. Guo, Y. Cao and J. Zhou, Preparation of high performance Pd catalysts supported on untreated multi-walled carbon nanotubes for formic acid oxidation, Electrochim. Acta, 2010, 55, 6036 CrossRef CAS PubMed.
- I. Katsounaros, W. B. Schneider, J. C. Meier, U. Benedikt, P. U. Biedermann, A. A. Auer and K. J. J. Mayrhofer, Hydrogen peroxide electrochemistry on platinum: towards understanding the oxygen reduction reaction mechanism, Phys. Chem. Chem. Phys., 2012, 14, 7384 RSC.
- I. Katsounaros, W. B. Schneider, J. C. Meier, U. Benedikt, P. U. Biedermann, A. Cuesta, A. A. Auer and K. J. J. Mayrhofer, The impact of spectator species on the interaction of H2O2 with platinum–implications for the oxygen reduction reaction pathways, Phys. Chem. Chem. Phys., 2013, 15, 8058 RSC.
- I. Katsounaros and K. J. J. Mayrhofer, The influence of non-covalent interactions on the hydrogen peroxide electrochemistry on platinum in alkaline electrolytes, Chem. Commun., 2012, 48, 6660 RSC.
- A. A. Topalov, S. Cherevko, A. R. Zeradjanin, J. C. Meier, I. Katsounaros and K. J. J. Mayrhofer, Towards a comprehensive understanding of platinum dissolution in acidic media, Chem. Sci., 2014, 5, 631 RSC.
- S. Cherevko, A. A. Topalov, A. R. Zeradjanin, I. Katsounaros and K. J. J. Mayrhofer, Gold dissolution: towards understanding of noble metal corrosion, RSC Adv., 2013, 3, 16516 RSC.
- F. Yang, K. Cheng, T. Wu, Y. Zhang, J. Yin, G. Wang and D. Cao, Au–Pd nanoparticles supported on carbon fiber cloth as the electrocatalyst for H2O2 electroreduction in acid medium, J. Power Sources, 2013, 233, 252 CrossRef CAS PubMed.
- F. Yang, K. Cheng, T. Wu, Y. Zhang, J. Yin, G. Wang and D. Cao, Dendritic palladium decorated with gold by potential pulse electrodeposition: Enhanced electrocatalytic activity for H2O2 electroreduction and electrooxidation, Electrochim. Acta, 2013, 99, 54 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |