
Iron(III) bromide catalyzed bromination of 2-tert-butylpyrene and corresponding position-dependent aryl-functionalized pyrene derivatives†
Xing Fengab,
Jian-Yong Hubc,
Hirotsugu Tomiyasub,
Zhu Taod,
Carl Redshawe,
Mark R. J. Elsegoodf,
Lynne Horsburghf,
Simon J. Teatg,
Xian-Fu Weia and
Takehiko Yamato*b
aBeijing Institute of Graphic Communication, Beijing 102600, P.R. China
bDepartment of Applied Chemistry, Faculty of Science and Engineering, Saga University, Honjo-machi 1, Saga 840-8502, Japan. E-mail: yamatot@cc.saga-u.ac.jp
cEmergent Molecular Function Research Group, RIKEN Center for Emergent Matter Science (CEMS), Wako, Saitama 351-0198, Japan
dKey Laboratory of Macrocyclic and Supramolecular Chemistry of Guizhou Province, Guizhou University, Guiyang, Guizhou 550025, P. R. China
eDepartment of Chemistry, The University of Hull, Cottingham Road, Hull, Yorkshire HU6 7RX, UK
fChemistry Department, Loughborough University, Loughborough, LE113TU, UK
gALS, Berkeley Lab, 1 Cyclotron Road, MS2-400, Berkeley, CA 94720, USA
First published on 4th December 2014
Abstract
The present work probes the bromination mechanism of 2-tert-butylpyrene (1), which regioselectively affords mono-, di-, tri- and tetra-bromopyrenes, by theoretical calculation and detailed experimental methods. The bromine atom may be directed to the K-region (positions 5- and 9-) instead of the more reactive 6- and 8-positions in the presence of iron powder. In this process, FeBr3 plays a significant role to release steric hindrance or lower the activation energy of the rearrangement. The intermediate bromopyrene derivatives were isolated and confirmed by 1H NMR spectrometry, mass spectroscopy and elemental analysis. Further evidence on substitution position originated from a series of aryl substituted pyrene derivatives, which were obtained from the corresponding bromopyrenes on reaction with 4-methoxy-phenylboronic acid by a Suzuki–Miyaura cross-coupling reaction. All position-dependent aryl-functionalized pyrene derivatives are characterized by single X-ray diffraction, 1H/13C NMR, FT-IR and MS, and offered straightforward evidence to support our conclusion. Furthermore, the photophysical properties of a series of compounds were confirmed by fluorescence and absorption, as well as by fluorescence lifetime measurements.
Introduction
Pyrene and its derivatives1 belong to a classical family of polycyclic aromatic hydrocarbons (PHAs) that have been extensively investigated for light-emitting device applications over recent years. This interest stems from their inherent chemical and photochemical characteristics, in particular an excellent deep blue chromophore which exhibits great chemical stability and high charge carrier mobility. However, owing to its planar structure, pyrene has a strong tendency to form π-aggregates/excimers, which, in-turn, leads to an excimer emission band and the quenching of fluorescence in condensed media and a resulting low fluorescence quantum yield. With this in mind, enormous effort has been paid towards exploring new methods to functionalize the pyrene-core for developing molecular materials and applications thereof.In general, due to the presence of nodal planes located at the 2- and 7-positions in both the highest occupied molecular orbital's (HOMO) and the lowest unoccupied molecular orbital's (LUMO) of pyrene, substituting pyrene at the 2- and/or 2,7-positions is more difficult compared to other positions (such as 1-, 3-, 6- and 8-positions (active site)).2 Thus, there are few examples which focus on substituting at the 2- and 7-positions of pyrene by borylation,3 bromination,4 nitration,5 oxidation6 and tert-butylation.7 On the other hand, the active sites, namely the 1-, 3-, 6- and 8-positions have been thoroughly examined and the products used in a variety of applications as optical materials.1a,1b Since we first reported8 the oxidation of pyrene at the K-region (4-, 5-, 9- and 10-positions) in 1997 by stepwise synthetic methods, the K-region also has been explored as a convenient synthetic route to the ketone9 and used for preparing pyrene-fused azaacene derivatives for application in organic semiconductors.10
As previously mentioned, bromopyrenes are significant intermediate compounds which play an important role in modern organic chemistry, not only for synthetic methodology, but also for advanced optoelectronic materials. Commonly, the 1-, 3-, 6- and 8-positions of pyrene preferentially undergo electrophilic aromatic substitution (SEAr) reactions. Therefore, mono-, bis-, tri- and tetrakis-substituted pyrenes were synthesized for organic electronic devices1 and fluorescence probes.11 For example, Thummel et al.12 discussed the crystal packing of 1,3-, 1,6-, 1,8- and 2,7-bis(2-[1,10]phenanthrolinyl)pyrenes and their use as pyrene-bridging ligands in ruthenium(II) chemistry, as evidenced by 1H NMR spectra and single-crystal X-ray crystallography. Sankararaman13 and co-workers reported a pyrene octaaldehyde derivative from 1,3,6,8-tetrabromopyrene, which can cause molecular aggregations in nonpolar solvents and in the solid-state through cooperativity of the intermolecular π–π stacking and C–H⋯O interactions, which has potential application in the field of molecular optoelectronics. Chow14 and co-workers synthesized sterically congested tetraaryl-pyrenes as efficient blue emitters that exhibited pure-blue electroluminescence and formed respectable organic light-emitting diodes (OLEDs). More recently, Konishi15 and co-workers systematically alkylated the active sites, namely the 1-, 3-, 6- and 8-positions of pyrene, and investigated the effects of the number of alkyl substituents on the photophysical properties of the pyrene chromophores. Recently, we reported a new type of fluorescent sensor based on a pyrene-linked triazole-modified hexahomotrioxa-calix[3]arene, which used the 1-pyrenyl moiety for selectively detecting Zn2+ and H2PO4− ions in neutral solutions.16
Bromination of the pyrene not only occurred at the active sites of 1-, 3-, 6- and 8-positions, but also substitution occurred at the K-region, namely the 4-, 5-, 9- and 10-positions. For instance, our group reported a series of pyrene-based cruciform/hand-shaped light-emitting monomers with highly emissive pure-blue fluorescence from tetrabromo/pentabromopyrene.17 Very recently, we explored a new bromide precursor, 1,3,5,9-tetrabromo-7-tert-butylpyrene,18 prepared via bromination of 2-tert-butylpyrene (1) in CH2Cl2 at room temperature using iron powder as the catalyst.
About twenty years ago, we reported an FeBr3-catalyzed rearrangement of a pyrene-based material in which the bromine atom was transferred from the active site (1-position) to the K-region (4-position), i.e. bromination of 2,7-di-tert-butylpyrene with 1.1 mole equiv. of bromine in the presence of iron powder to afford 1-bromo-2,7-di-tert-butylpyrene, which can be further brominated with excess bromine in the presence of FeBr3, and afforded 4,5,9,10-tetrabromo-2,7-di-tert-butylpyrene (Scheme 1).8 However, the detailed bromination mechanism still remains unclear.
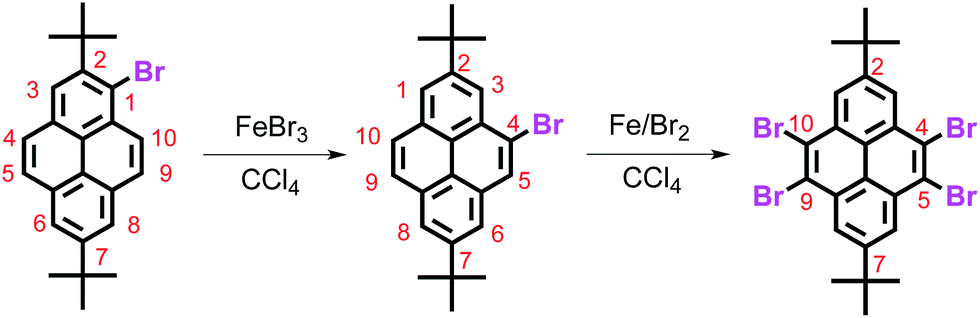 | ||
| Scheme 1 FeBr3-catalyzed rearrangement to afford 4,5,9,10-tetrabromo-2,7-di-tert-butylpyrene. | ||
Interestingly, we recently succeeded in developing the bromide precursor 1,3,5,9-tetrabromo-7-tert-butylpyrene from 2-tert-butyl-pyrene (1) using iron-powder catalysis.18 We speculated that the sequence of reactions involved a stepwise bromination process from the 1-, 3-, 5- to the 9-position. In this case, it seemed easy to understand that the bromination reactions of the pyrene preferentially occurred at the active sites of the 1- and 3-positions owing to the tert-butyl group protecting the ring against electrophilic attack at the 6- and 8-positions.19 Following this, as expected, the next step would be to substitute regioselectively at the 5- and 9-positions, which should be favoured given that the bromine atoms substituted at the 1- and 3-positions would sterically hinder the 4- and 10-positions. However, further experimental results revealed that the reaction process was more complicated than our initial predictions.
It is well known that bromination reactions can be violent and complex processes, thus to investigate the bromination mechanism is far from simple, because it is difficult to capture the transition state or a transition state analogue by experimental methods. Given this, the mechanism of electrophilic substitution was generally investigated by kinetic and stereochemical studies, or by theoretical analyses.20 In additional, if the experimental conditions cannot be well controlled, then the final components will be complicated and hard to characterize.21 In this paper, we present the first example of the systematic exploration of the bromination rearrangement reaction of pyrene affording the mono-, di-, tri- and tetra-bromopyrenes by detailed experimental procedures and theoretical calculation. The corresponding positions-dependent arylpyrenes derivatives containing the 4-methoxyphenyl group were synthesized from the corresponding bromopyrenes via a Suzuki–Miyaura cross-coupling reaction, which was characterized by single crystal X-ray diffraction and 1H NMR spectroscopy. The detailed results strongly supported the bromo-substituted positions in the pyrene ring as per the previous assumption. Furthermore, the effects of the methoxyphenyl group (both number and the substitution pattern) on their photophysical properties, as well as for the molecular packing, were investigated.
Results and discussion
Stoichiometric bromination of 2-tert-butylpyrene (1)
A three-neck round bottom flask fitted with a dropping funnel and a CaCl2 drying tube, was filled with 1 (200 mg, 0.78 mmol) in 20 mL of CH2Cl2 and was stirred for 30 min. at 0 °C. A solution of Br2 (depending on stoichiometric ratio) in 5 mL of CH2Cl2 was added drop-wise. After the addition of bromine was completed, the mixture was warmed to room temperature (28 °C) and stirred for 5 h. The crude product was washed with hot hexane and the yields of products are compiled in Table 1.| Run | Substrate | Reagents | Reagents/1 (mole rate) | Products (%) |
|---|---|---|---|---|
| a The isolated yields are shown in bracket.b Yields were determined by 1H NMR analysis and shown in parentheses. | ||||
| 1 | 1 | Br2–Fe | 1.0 | 2a [83] |
| 2 | 1 | BTMABr3 | 1.0 | 2a [84] |
| 3 | 1 | Br2–Fe | 2.0 | 2a [50], 2b [35] |
| 4 | 1 | BTMABr3 | 3.5 | 2b [76] |
| 5 | 1 | Br2 | 3.9 | 2c [65] |
| 6 | 1 | Br2–Fe | 3.0 | 2d (25), 2f (50)b |
| 7 | 1 | Br2–Fe | 6.0 | 2f [84] |
| 8 | 2c | Br2–Fe | 2.5 | 2e (70), 2f (30)b |
| 9 | 2e + 2f | Br2–Fe | 2.9 | 2e (25), 2f (75)b |
The synthesis of the key intermediate bromopyrenes is shown in Scheme 2. 1-Bromo-7-tert-butylpyrene (2a) and 1,3-dibromo-7-tert-butylpyrene (2b) have been synthesized with two kinds of bromination reagents and were then used to achieve the chemical modifications of the pyrenes for the required products.19 Firstly, (1) a mixture of 1 and 1.0 equiv. bromine in CH2Cl2 at 28 °C in the presence of an iron powder catalyst, afforded 2a in 83% yield; on the other hand, in the absence of iron powder, a mixture of 1 and 1.0 equiv. bromine in CH2Cl2 at −78 °C afforded 2a in 75% yield;19b (2) a mixture of 1 and BTMABr3 (1.0 equiv.) in CH2Cl2 at 28 °C afforded the desired product 2a in 84% yield. (3) According to the literature,19b a mixture of Br2 and 1 in anhydrous CH2Cl2 at −78 °C afforded 2a in 89% yield in the absence of iron powder; however, in the presence of iron powder, a mixture of 1 and 2.0 equiv. of bromine at 28 °C afforded a mixture 2a in 50% yield and 2b in 35% yield. (4) A mixture of 1 and BTMABr3 (3.5 equiv.) in CH2Cl2 at 28 °C afforded the desired product 2b in 76% yield. Similarly, (5) for comparison, we synthesized 1,3,6-tribromo-7-tert-butylpyrene (2c) in the absence of iron powder in 65% yield according to the reported procedure.22 (6) When 1 was mixed with stoichiometric Br2 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3–5) under the same experimental conditions, the intermediate product 1,3,5-tribromo-7-tert-butylpyrene (2d) was obtained with 2f, which could not be separated from the crude compound by column chromatography. We attempted to isolate both 2d and 2f in pure form by high-performance liquid chromatography (HPLC) but failed. (7) When the same reaction was carried out with 6.0 equiv. of bromine in the presence of iron powder, the Lewis acid-catalyzed rearrangement of bromine was observed to give 1,3,5,9-tetrabromo-7-tert-butylpyrene 2f in 84% yield.18 It seems that compound 2f might be formed by the isomerization of compound 2c under FeBr3-catalyzed conditions, which should be produced from bromine and iron powder present during the bromination. (8) Reaction of 2c with Br2 (1:2.5) was carried out in the presence of iron powder. A mixture of the bromides 1,3,5,8-tetrabromo-7-tert-butylpyrene 2e and 1,3,5,9-tetrabromo-7-tert-butylpyrene 2f was obtained in the ratio of 7:3 (determined by their 1H NMR spectra analysis); (9) when a mixture of 2e and 2f was treated with 2.5 equiv. of bromine in the presence of iron powder in CH2Cl2 solution at 28 °C for 8 h, the expected product, 2f was obtained in 75% yield. (10) However, attempted isomerization of compound 2e to 2f with other Lewis acids, such as TiCl4, AlCl3 or FeCl3, performed under the same reaction conditions failed; only the starting compound 2e was quantitatively recovered. Brominations of compound 1 in the presence of iron powder to afford the bromo-substituted pyrene derivatives 2 were carried out under various reaction conditions and the detailed results are summarized in Table 1.
:3–5) under the same experimental conditions, the intermediate product 1,3,5-tribromo-7-tert-butylpyrene (2d) was obtained with 2f, which could not be separated from the crude compound by column chromatography. We attempted to isolate both 2d and 2f in pure form by high-performance liquid chromatography (HPLC) but failed. (7) When the same reaction was carried out with 6.0 equiv. of bromine in the presence of iron powder, the Lewis acid-catalyzed rearrangement of bromine was observed to give 1,3,5,9-tetrabromo-7-tert-butylpyrene 2f in 84% yield.18 It seems that compound 2f might be formed by the isomerization of compound 2c under FeBr3-catalyzed conditions, which should be produced from bromine and iron powder present during the bromination. (8) Reaction of 2c with Br2 (1:2.5) was carried out in the presence of iron powder. A mixture of the bromides 1,3,5,8-tetrabromo-7-tert-butylpyrene 2e and 1,3,5,9-tetrabromo-7-tert-butylpyrene 2f was obtained in the ratio of 7:3 (determined by their 1H NMR spectra analysis); (9) when a mixture of 2e and 2f was treated with 2.5 equiv. of bromine in the presence of iron powder in CH2Cl2 solution at 28 °C for 8 h, the expected product, 2f was obtained in 75% yield. (10) However, attempted isomerization of compound 2e to 2f with other Lewis acids, such as TiCl4, AlCl3 or FeCl3, performed under the same reaction conditions failed; only the starting compound 2e was quantitatively recovered. Brominations of compound 1 in the presence of iron powder to afford the bromo-substituted pyrene derivatives 2 were carried out under various reaction conditions and the detailed results are summarized in Table 1.
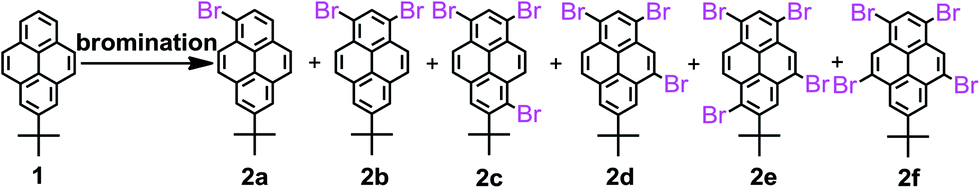 | ||
| Scheme 2 Bromination of 2-tert-butylpyrene for 2. | ||
Regioselective bromination mechanism of 2-tert-butylpyrene (1)
From the previous experimental sections, 2a, 2b, and 2c were prepared from reactions that do not depend on the presence of iron(III) bromine, rather, the stoichiometric ratio of bromine reagent. Process is a classical electrophilic substitution reaction. This result is strongly attributed to the high reactivity of the 1-, 3-, 6- and 8-positions in the pyrene ring. However, in the following bromination reactions for 2d and 2e the role played by the FeBr3-induced rearrangement to form the 2f is not clear.To disclose the bromination mechanism of 1, density functional theory (DFT) calculations (B3LYP/6-31G* basis set) were carried out using the Gaussian 03 software package for investigating the potential-energy surface of each bromopyrene derivative in Scheme 3; B3LYP function was chosen. The geometric structures of bromo-substituted compounds 2 are optimized at 6-31G* level and their parameters are given in ESI.† The frequency and the electronic distribution of 2 were further tested, the results show that the vibration frequency of 2 are with positive value, without imaginary frequency, indicating that the calculation of the molecular structure energy is at the minimum and stable. The relative free energy (ΔG298) is exergonic by 7.9 kcal mol−1 in the process of bromination of 1, leading to 2a, which can be further bromination to afford 2b and 2c with an exergonic reaction of 8.2 kcal mol−1 and 15.4 kcal mol−1, respectively.
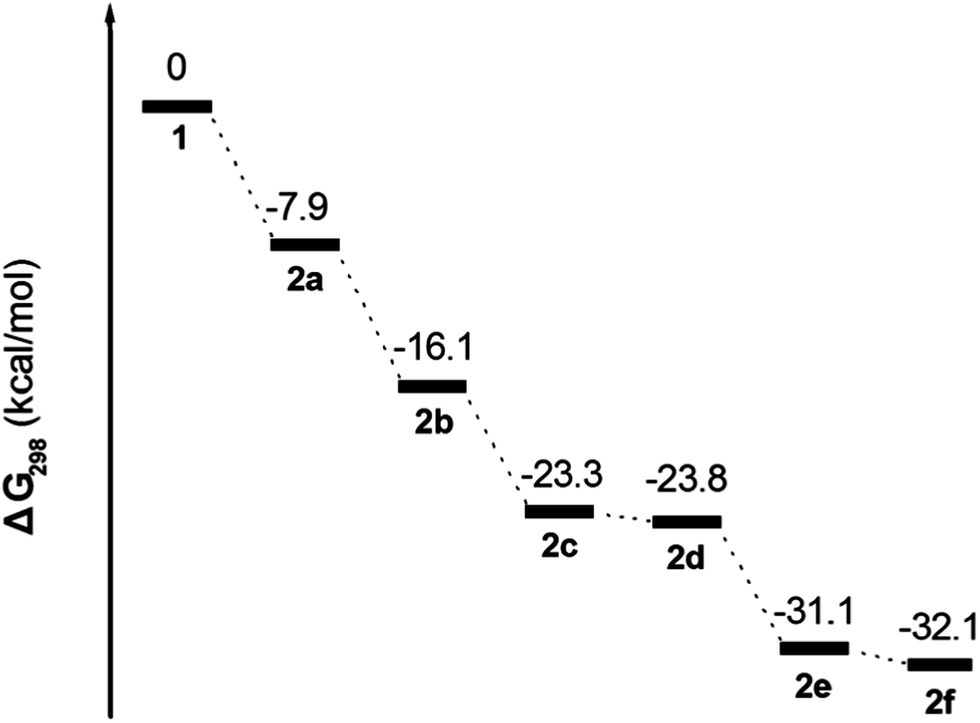 | ||
| Scheme 3 Potential energy surface for 1 and 2. (Gaussian 03W (B3LYP/6-31G* basis set)). | ||
Reaction of 1 with the bromine reagent leads to 2c with an exergonic reaction of 23.3 kcal mol−1, whilst the subsequent bromination steps from 2c or 2d to 2e and 2f proceed with an exergonic is ≈8.8 kcal mol−1.
Comparison of the geometric structures of 2, shows that the angle of the bromine atom at the 6-position of 2c (∠Br3–C5–C6 = 115.5°) or 8-position of 2e (∠Br4–C8–C7 = 115.7°), is less than in other bromo substituted-positions (for example: ∠Br1–C1–C2 = 120.6 in 2a). The tert-butyl group located at the 7-position of the pyrene core is slightly crowded in 2c and 2e with an angle of 125.7° ± 0.1, which is larger than those in 1 and 2a–b, 2d and 2f. Interestingly, 2c or 2d was generated from 2b with similar exergonic (7.2 kcal mol−1 for 2c and 7.7 kcal mol−1 for 2d). However, 2d cannot be observed by bromination of 2b with an excess of bromine without iron powder present in our experiment. In fact, in the presence of iron(III) bromide, 2a–c and 2e was synthesized via Friedel–Crafts-type reactions from 1 with bromine. From Scheme 3, the presence of iron(III) bromide might contribute to the lowering of the activation energy of bromination and inducing intramolecular bromine rearrangements in the process for 2d and 2f, given that the bromine atom would shift from the active site (6- or 8-position) to the K-region (5- or 9-position).
The possible bromination reaction pathway has been summarized in Scheme 4. Firstly, the relatively easy electrophilic substitution at the ortho-position to a tert-butyl group (6- or 8-position) on the pyrene ring is remarkable because usually the steric bulkiness of a tert-butyl group might be expected to direct the substitution towards other positions of the pyrene ring.7,8 This result is strongly attributable to the high reactivity of the 1-, 3-, 6- and 8-positions of the pyrene ring. Second, however, the pyrene exhibits a special electronic structure that of the Kekulé structure (I, II and III); the greatest number of benzenoid rings should have the greatest weight in the superposition diagram. Meanwhile, this also shows that the greatest number of double bonds in the “exposed” position has the greatest weight (see ESI†).23 Thus, when the bromide is attracted the 6-position of 2b, due to the steric strain involving the tert-butyl group and lowering of the Gibbs free energy, the bromide would rearrange to the 5-position under FeBr3-catalysis and afford 2c. Similarly, the tetrabromide 2f was obtained by the same FeBr3-catalyzed rearrangement in the bromination of tribromopyrenes 2c and 2d in the presence of iron powder. The above results strongly suggest that compounds 2c, 2d and 2e were the intermediates for the formation of the 1,3,5,9-tetrabromo-7-tert-butylpyrene 2f.
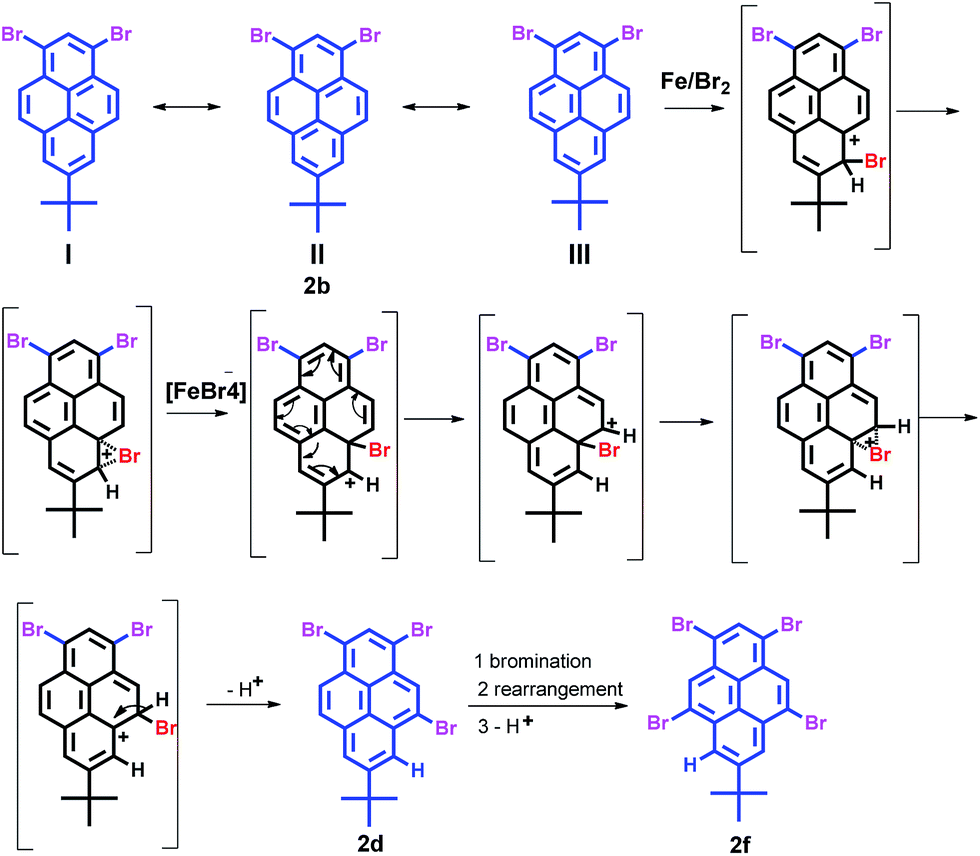 | ||
| Scheme 4 Possible regioselective bromination mechanism of 1. | ||
In this reaction, iron powder leads to the title products where bromine is directed to the K-region (positions 5- and 9-) instead of the more reactive positions 6- and 8-. Two reasons can explain this phenomenon of FeBr3-induced intramolecular bromine rearrangement: one possible reason is a release of steric hindrance as the driven force for this rearrangement, which is also possible in the absence of FeBr3 where no rearrangement is observed. Another possible reason from DFT calculations is a lowering of the activation energy of the rearrangement induced by FeBr3.
In order to further investigate bromo-substitution positions of the pyrene in more detail, a series of aryl-substituted pyrenes (3) were prepared from the resultant bromopyrenes using 4-methoxyphenylboronic acid via a Pd-catalyzed cross-coupling reaction. Using the 4-methoxyphenyl group as an effective substituent, several different shaped pyrenes 3 were synthesized and are displayed in Scheme 5: 7-tert-butyl-1-(4-methoxyphenyl)pyrene (3a), 7-tert-butyl-1,3-bis(4-methoxyphenyl) pyrene (3b),19a 7-tert-butyl-1,3,6-tris(4-methoxyphenyl)pyrene (3c), 7-tert-butyl-1,3,5-tris-(4-methoxyphenyl)pyrene (3d), 7-tert-butyl-1,3,5,8-tetrakis(4-methoxyphenyl)pyrene (3e) and 7-tert-butyl-1,3,5,9-tetrakis(4-methoxy)phenylpyrene (3f).18 The molecular structures of all were characterized by their 1H/13C NMR spectra, single-crystal X-ray diffraction, FT-IR spectroscopy, mass spectrometry, as well as by elemental analysis. All analysis results herein on the aryl-substituted pyrene 3 strongly supported our previous predictions.
 | ||
| Scheme 5 Synthetic route for compound 3 via a Suzuki–Miyaura cross-coupling reaction, reagents and conditions: 4-methoxylphenylboronic acid, [Pd(PPh3)4], Na2CO3, toluene/EtOH, 24 h, 90 °C. | ||
The performance of the organic compounds in optoelectronic devices strongly relies on the molecular packing and intra/intermolecular interactions in the solid-state. Therefore, investigating the effects of a structure–property relationship between the substituent groups on the pyrene-core using crystal structure and photophysical properties is significant for organic electroluminescence materials. Herein, we expected that the integration of poly-methoxyphenyl groups with the pyrene core in a molecular structure might influence the crystal packing, leading to favourable optical features and charge-transport properties that could be useful in optoelectronic devices.
Description of crystal structures
Previously, we reported that several Y-shaped aryl-substituted pyrenes with electron-donating/withdrawing groups at the para-position of the C6H4 rings inefficiently impact on the molecular packing in the solid-state.19a Konishi et al. have validated that alkyl groups located at the 1-, 3-, 6- and/or 8-positions of the pyrene ring play significant roles in tuning the photophysical properties.15 Herein, this article presents a series of position-substituted pyrenes that not only take place at the active sites (1-, 3-, 6- and 8-positions) but also the K-region (4- and 9-positions) of pyrene, and allow us to shed light on the effect of multiple 4-methoxyphenyl groups on the molecular packing and optical physical properties.Crystals of 3 suitable for X-ray structure analysis were grown from mixed solvents via slow evaporation at room temperature. The key crystallographic data are summarized in ESI Table S2;† the crystal structures of molecules for 3 are shown in Fig. 1.
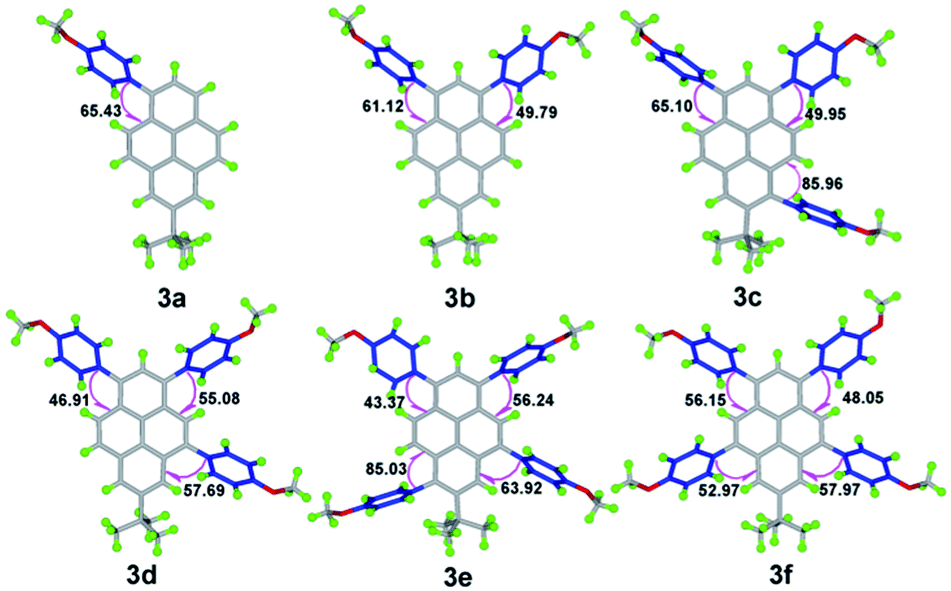 | ||
| Fig. 1 X-ray structures of compounds 3. | ||
Colourless crystals of compound 3a suitable for X-ray crystallographic analysis were obtained by crystallization from a mixture of dichloromethane and hexane (1:2, v/v). The single-crystal X-ray structure is depicted in Fig. 2. It can be seen that the 4-methoxyphenyl groups in this molecule form a torsion angle (65.43 (17)°) with the plane of the central pyrene ring to prevent face-to face π-stacking and steric clashes between ortho H atoms on the phenyl ring and those at the 3- and 9-positions. An interesting feature of the compound in the solid-state is that there is an intermolecular C–H⋯π interaction (C19–H19⋯C3 = 2.88 Å, C21–H21⋯C7 = 2.85 Å, C23–H23⋯C13 = 2.88 Å) between neighbouring molecules. These interactions led to a comparatively large twist angle between the pyrene core and the methoxyphenyl fragment, and effectively suppress the formation of the π–π stacking.
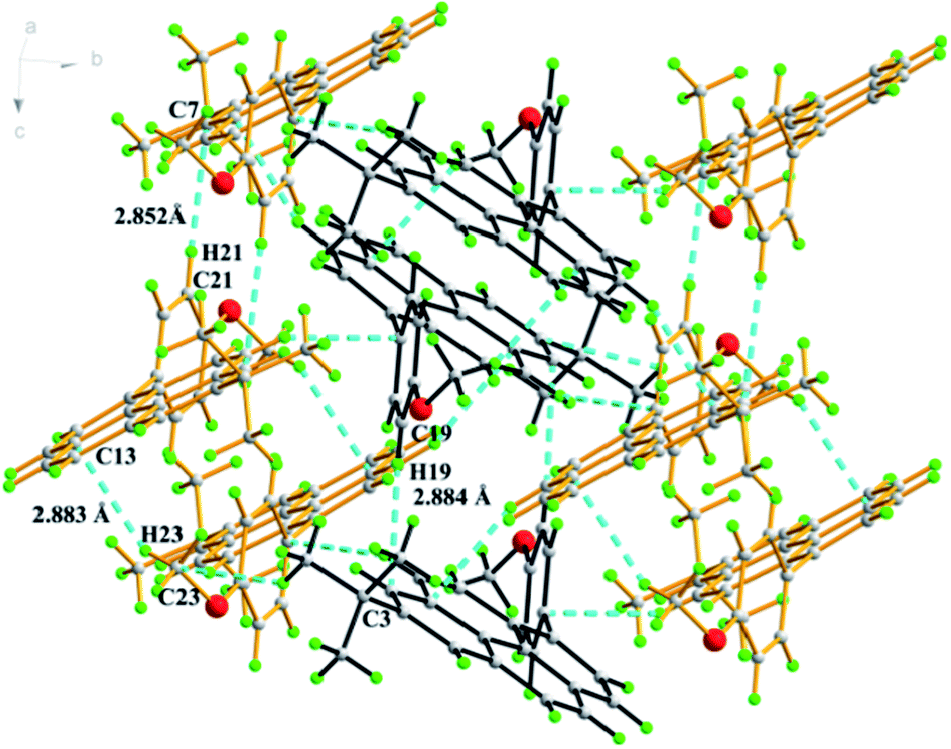 | ||
| Fig. 2 Crystal packing of 3a by C–H⋯π interactions. | ||
On increasing the numbers of 4-methoxyphenyl groups and substituting at different positions in the pyrene derivatives 3a → f, the space groups (orthorhombic for 3a, monoclinic for 3b, 3c and 3e, triclinic for 3d and 3f) became more asymmetric. It can be seen that the torsional angles between the 4-methoxyphenyl groups and the pyrene core decreased from 65.43 (17)° to 48.08 (18)°. With the number of substituted groups increasing, the molecular conformations tend towards increasing co-planarity.24 However, without the tert-butyl group located at the nodal planes involving the 2-/7-positions in 1,3,6,8-tetrakis(4-methoxyphenyl)pyrene (4),18 the torsion angle is unexpectedly larger than 3 and is up to 76.1 (4)°.
Colourless rod crystals of 3c suitable for X-ray diffraction were recrystallization from a mixture of dichloromethane and methanol by slow evaporation at room temperature. Fig. 3 shows the crystal packing in 3c. The crystal structure revealed the novel asymmetric substitution of the pyrene core. As expected, the 4-methoxyphenyl group successfully substituted at the 6-position of the pyrene with an approximately perpendicular torsion angle of 88.58(4)°. In the crowded region at the 6- and 7-positions, two bulky moieties were introduced, which contact each other by steric interaction. This causes two-fold tert-butyl group disorder with an occupancy ratio 0.634 (12):0.366 (12) for C18, C19 and C20 and an intramolecular short contact (C18–H18B⋯C26 = 2.81 Å) and significant distortion for the Csp2 hybridization with an angle of 123.81(15)° at C4–C5–C17. In the off-set packing system of 3c, the two proximal pyrene molecular planes have a long centroid-centroid distance of 7.97 Å and no π⋯π stacking was observed. The adjacent molecules interacted by a classical C–H⋯π bond (C34–H34⋯C1 = 2.84 Å, C32–H32⋯C15 = 2.82 Å, C23–H23⋯C10 = 2.84 Å). It seems that the close proximity of the 4-methoxyphenyl moiety and the tert-butyl group caused strong intermolecular steric hindrance in this packing system.
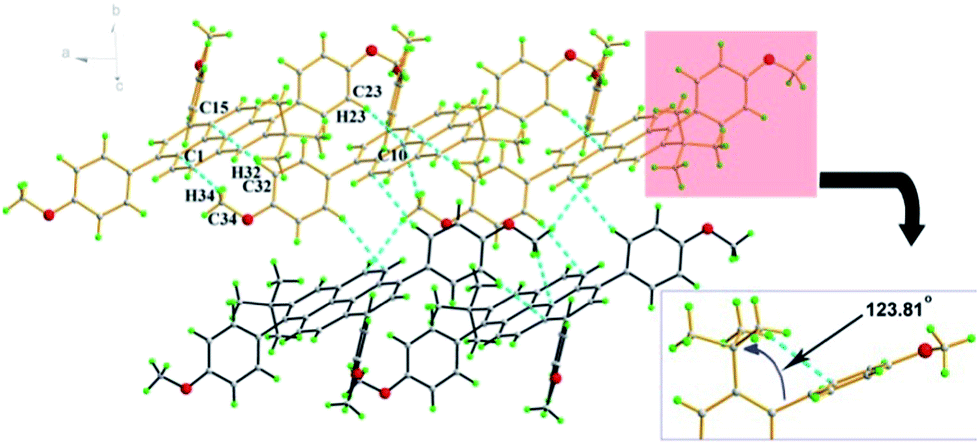 | ||
| Fig. 3 Crystal packing in 3c by C–H⋯π interactions. | ||
Yellow needles of 3d were obtained from a mixed solution of dichloromethane–hexane = 1:1; the asymmetric unit of compound 3d contains two molecules (Fig. 4). The molecule exhibits non-planarity and the central pyrene ring has a slight bend; possibly arising from the imbalance of the electrostatic potential on the molecular surface.24 The inter-planar angles between the central pyrene core and the outer substituent phenyl rings range from 44.2(5)° to 57.7(5)°. The crystal structure was arranged in columns along the a-axis, through essentially parallel, or near-parallel interactions between translationally equivalent molecules. Each molecule is interlaced with adjacent columns along the a-axis by the formation of π–π stacking with a centroid-to-centroid distance of 4.07 Å and an inter-planar angle of 0°. There are numerous C–H⋯π interactions formed between phenyl hydrogens and neighbouring aromatic rings.
 | ||
| Fig. 4 Crystal packing in 3d by C–H⋯π interactions. | ||
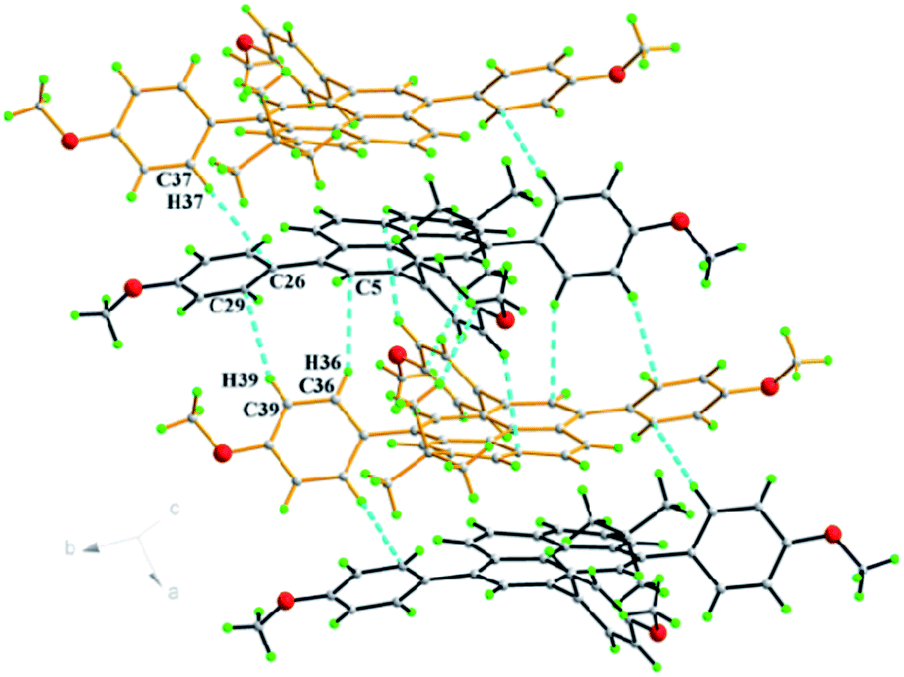
Compound 3e was dissolved in CHCl3 and kept in a CH3OH atmosphere at room temperature to afford orange single crystals suitable for X-ray diffraction. The crystal system of 3e is monoclinic with space group P21/c and is shown in Fig. 5. A similar arrangement pattern to 3d was observed. The molecules also adopt a slipped face-to-face and π–π stacking pattern along the b-axis with a centroid-to-centroid distance of 4.96 Å. This is longer than that observed in 3d, arising from an extra 4-methoxyphenyl moiety playing a role to prevent the neighbouring molecules getting too close to each other. Also, the 4-methoxyphenyl group and bulky tert-butyl group share a crowded space at the 1- and 2-positions of the pyrene, and the sterics result in the Csp2 hybridization angle C4–C5–C17 changing to 124.07(6)° via intramolecular hydrogen bonds (C19–H19B⋯C26 = 2.56 Å, C18–H18B⋯C22 = 2.53 Å).
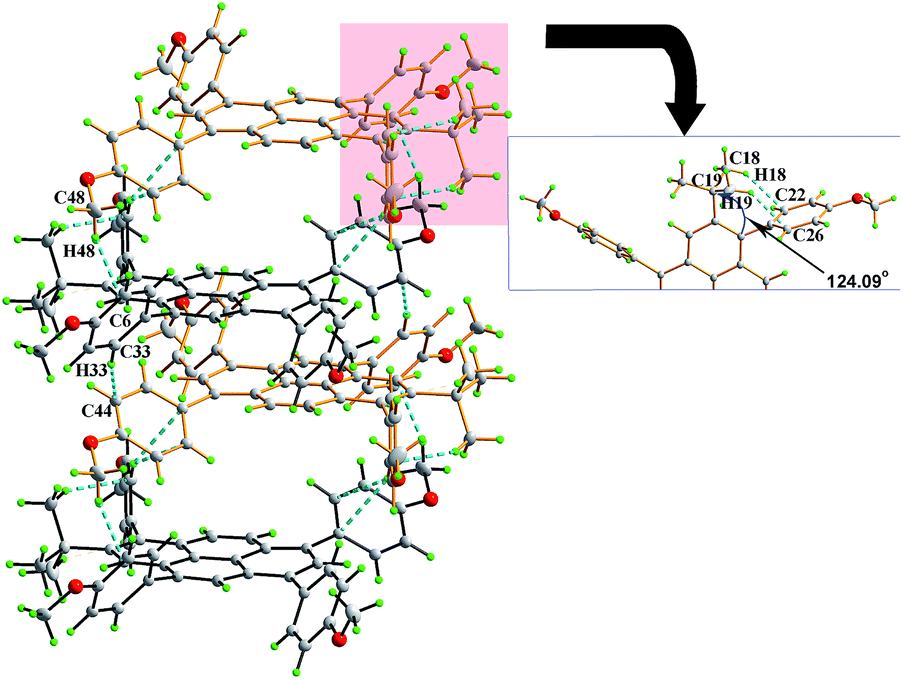 | ||
| Fig. 5 (a) Crystal packing in 3e by C–H⋯π interactions. | ||
Comparison of the tetra-substituted pyrenes 3e, 3f and 1,3,6,8-tetrakis(4-methoxyphenyl)pyrene (4), reveals that the crystal packing in the solid-state and is attributed to the position-substituted diversification.18,25 For the packing structures of 3a to 3f, the X-ray diffraction revealed that the torsion angle decreased between the substituent group and the pyrene core and the molecular packing varied significantly from 3D (column structure) to 2D (planar structure) when the number of 4-methoxyphenyl groups increased. As mentioned above, the number of substituent groups and their positions, as well as the bulky tert-butyl groups play an important role in arranging the molecular conformations. The structures became more planar, which in turn was beneficial for π-conjugation and improved the optical density, leading to strong FL emission in the solid-state.26 In the next section, we discuss the resulting photophysical properties resulting from the presence of the multiple 4-methoxyphenyl substituents.
Photophysical properties
The normalized UV-vis absorption and fluorescence spectra for 3 recorded in dichloromethane are shown in Fig. 6, for comparison, 1,3,6,8-tetrakis(4-methoxyphenyl)pyrene (4)18 and 2-tert-butyl-4,5,7,9,10-pentakis(4-methoxyphenyl)pyrene (5)17a are summarized here for investigating the effect of the positions-dependent aryl-functionalized pyrene derivatives for the packing structures and photophysical properties, and the corresponding photophysical data is summarized in Table 2. Except for 3c and 5, all molecules exhibited very similar photophysical characteristics with well-resolved spectral bands in both the short wavelength of 283–305 nm and long wavelength of 347–391 nm regions. The absorption spectra of 3 → 4 show a maximum band at 347 nm for 3a, 363 nm for 3b, 363 nm for 3d, 381 nm for 3e, 381 nm for 3f and 391 nm for 4. However, 3c has two absorption peaks centered at 295 nm and 375 nm with a shoulder peak at 363 nm; similarly, 5 displays two maximum absorption peaks at 296 nm and 356 nm with a shoulder peak at 343 nm. Clearly, both the number of substituents and the substitution position (pathway) strongly influence the electronic absorption;2 the absorption maximum of 3 revealed a remarkable red-shift. It is thought that such phenomena arise from the increasing number of peripheral arms in this series, which extend the π-conjugation of the pyrenes.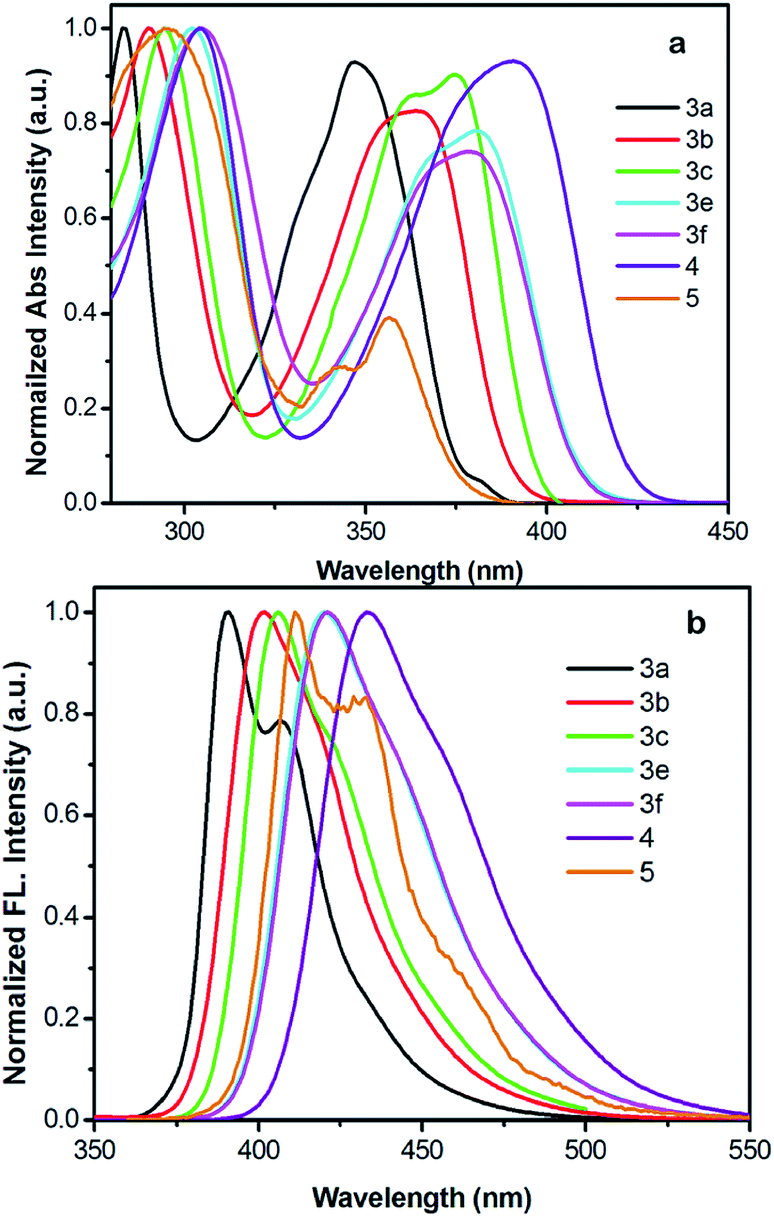 | ||
| Fig. 6 (a) Normalized UV-vis absorption and (b) emission spectra of compounds 3, 4 and 5 recorded in dichloromethane at ca. ∼10−5–10−6 M at 25 °C. | ||
| Compounds | λmaxabs (nm) solnsa/filmsb | λmax PL (nm) solnsa/filmsb | Φfc solns/films | HOMO (eV) | Energy gap (eV) | τa (ns) | Tmg/Tdh (°C) | ||
|---|---|---|---|---|---|---|---|---|---|
| a Measured in dichloromethane at room temperature.b Measured in thin films.c Measured in dichloromethane and in thin films, respectively.d Calculation by DFT (B3LYP/6-31G*).e Calculated from Onset of first oxidation potential according to equations: −(4.8 + Eonsetox).f Estimated from UV-vis absorption spectra in solution.g Melting temperature (Tm) obtained from differential scanning calorimetry (DSC) measurement.h Decomposition temperature (Td) obtained from thermogravimetric analysis (TGA). nd. No determination. | |||||||||
| 3a | 347 | nd | 391, 407 | nd | 0.41/nd | −5.06d/nd | 3.65d (3.30)f | 8.6 | 141/282 |
| 3b | 363 | 372 | 402 | 450 | 0.56/0.58 | −4.93/−5.44e | 3.51 (3.17) | 8.9 | 167/173 |
| 3c | 363, 375 | 381 | 406 | 471 | 0.71/0.28 | −4.84/−5.06 | 3.46 (3.13) | 5.2 | 249/nd |
| 3e | 381 | 388 | 420 | 441 | 0.76/0.58 | −4.76/−5.36 | 3.37 (3.03) | 2.2 | 240/399 |
| 3f | 379 | 369 | 421 | 443 | 0.90/0.72 | −4.76/−5.37 | 3.40 (3.03) | 5.8 | 330/410 |
| 4 | 391 | 405 | 434 | 488 | 0.94/nd | −4.71/nd | 3.24 (2.94) | 1.9 | 271/nd |
| 5 | 343, 356 | 357 | 411, 433 | 414 | 0.24/0.17 | −4.93/nd | 3.70 (3.31) | 18.8 | nd/433 |
The longest absorption (λmax) exhibited by 3e and 3f was approximately red shifted by ca. 34 nm versus 3a, indicating the HOMO–LUMO energy gap of 3 decreased on increasing the conjugation length. Interestingly, for the hand-shaped compound 5, despite it having the most substituents, the absorption does not show a significant red-shift in comparison with those of the tetrasubstituted derivatives 3e and 3f, which is probably a result of the nodal planes passing through the 2,7-positions, leading to a lower electronic density by the rotation of 4-methoxyphenyl group located at 7-position of pyrene core. This suggests the 2,7-substitutions have a small influence on the electronic interaction,2 and the results have also been reinforced by DFT calculations (mentioned below).
For the emission spectra, all compounds exhibit intense emissions in the blue region (391–434 nm). The emission maxima of 3 and 4 are bathochromically shifted depending on the numbers of 4-methoxyphenyl units, revealing an identical trend to their absorption spectra. No characteristic excimer fluorescence was observed in any of the spectra. It is worth noting that 1,3,5,8-functionalized pyrene 3e exhibits an emission with λem values of 420 nm, which almost overlaps with the emission spectrum (421 nm) of 1,3,5,9-functionalized pyrene (3f). In general, the substitution pattern of the pyrene moiety has a substantial effect on the fluorescence wavelength, and the effect of being substituted at the active 1-, 3-, 6-and 8-positions for the S1 ← S0 excitations is more significant than at the K-region (4-, 5-, 9- and 10-positions).2
In the case of 3e and 3f, smaller red-shifts in their electronic absorption profiles indicate that the structural changes and/or electronic distribution changes can cause an electronic communication missing in the pyrene cores.27 For 5, a deep-blue emission was observed with a maximum peak at 411 nm and a shoulder at 430 nm in solution.
The UV-vis absorption and emission spectra of selected pyrenes in the solid-state are shown in Fig. 7, and the optical data are summarized in Table 2. Compared with the corresponding solutions, the absorption for 3b, 3c, 3e, 4 and 5 films reveals a slight red-shift (about 10 nm) (Table 2). However, the absorption of 3f as a film shows a slight blue-shift in comparison with that in solution. This unusual blue-shift might be due to the different dielectric constant.28
 | ||
| Fig. 7 (a) Normalized UV-vis absorption and (b) emission spectra of 3 and 5 in thin films. | ||
The emission maxima of 3b, 3e and 3f as thin films exhibited red-shifts of less than 48 nm relative to those in solution, and the compound 3c exhibits a red-shift of 65 nm (from 406 nm to 471 nm) (Table 2). On increasing the numbers of 4-methoxyphenyl moieties, the red-shift decreased in the following order: 5 (3 nm) < 3f (22 nm) ≈ 3e (21 nm) < 3b (48 nm) < 4 (54 nm) < 3c (65 nm), indicating the positions of the substituents of the aryl-functionalized would influence the electronic interaction. Owing to the strong intermolecular interactions in the thin film of 3c and 4, the emission maxima exhibited a greater red-shift as a thin film state with high noise level.29
In contrast to the 1,3,6,8-tetrakis(4-methoxyphenyl)pyrene 4,18 that tends to exhibit a high noise level PL spectrum in the solid-state. the tetra-substituted pyrenes 3e and 3f exhibited clear and sharp emission peaks in the blue-region without extra excimer emissions in the solid-state owing to the bulky tert-butyl group located at the 7-position of the pyrene ring suppressing the aggregation. Compound 4 also presents a very high fluorescence quantum yield (Φcf) of the order of ∼0.94 in solution.
For comparison, the quantum yields of 3b, 3c, 3e and 3f in the solid-state were also investigated (0.58 for 3b, 0.28 for 3c, 0.58 for 3e and 0.72 for 3f). However, for 5, low fluorescence quantum yields in both solution and the solid-state were obtained due to energy loss that is likely occurring during the exciton migration.30 The fluorescence lifetime of 3a–c, 3e, 3f, 4 and 5 are 8.6 ns, 8.9 ns, 5.2 ns, 2.2 ns, 5.8 ns, 1.9 ns, 18.8 ns, respectively. Excellent optical features were obtained in these compounds, which make them of potential use in new optoelectronic devices, such as blue emitters in OLEDs, or as models for further exploring a new generation of organic materials based on pyrene.
Quantum chemistry computation
To gain further insight into the effect of multi-substituents and pathways on the electronic structure and spectral properties of compounds 3, 4 and 5, quantum chemical calculations were calculated using DFT methods at the B3LYP/6-31G* level. The calculated energies of the frontier molecular orbitals are presented in Fig. 8 and in the ESI.†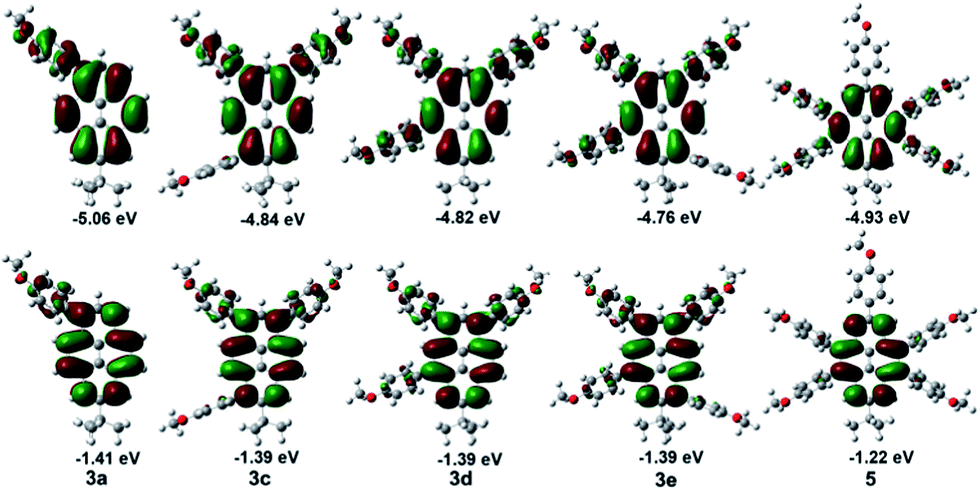 | ||
| Fig. 8 Computed molecular orbital plots (B3LYP/6-31G*) of compounds of 3 and 5; the upper plots represent the HOMOs, and the lower plots represent the LUMOs. | ||
Scrutiny of the electronic structures reveals that both HOMOs and LUMOs of 3 were primarily delocalized over the entire pyrene component, as well as slightly in the peripheral phenyl moiety, the only difference being in the energy of these frontier molecular orbitals, which in turn relied on the system architecture. For instance, on increasing of the numbers of the substituents from 3a to 3f, the HOMO values are more positive varying from −5.06 eV (3a) to −4.76 (3f) eV, the increased position-dependent substitution resulted in a lowering of both the HOMOs and LUMO by 0.3 eV and 0.05 eV, respectively. The effects of multiple substituents is greater for the HOMOs than for the LUMOs with a sizable shrinking of the HOMO–LUMO gap by 0.28 eV with respect to 3a, which is in good agreement with the experimentally measured results. The slight differences to those obtained by UV-vis absorption (ΔEgap opt = 0.27 eV) are due to the DFT-calculation being performed in the gas phase.
Compared with 3e/3f and 4, the presence of the tert-butyl group at the 7-position, could lead to a lower energy gap by lowering the molecular LUMOs. With the number of substituent groups increased, from the mono-substituted 3a to the tetra-substituted pyrenes 4 and 3e/3f, the energy gap of the representative molecules decreased. The special electronic structure of the penta-substituted pyrene 5 inhibits absorption spectra which is different form the others, due to the substituent group at the K region (4,5,9,10-positions) which favours a blue shift and improves the energy gap of the molecular structure. The 4-methoxyphenyl group when located at the nodal planes would weaken the electronic coupling over the entire pyrene. This has little influence on the S2 ← S0 absorption, but a larger influence on the S1 ← S0 absorption.31 So, These conclusions are also consistent with our quantum chemical calculations.
Electrochemistry
The electrochemical properties of selected compounds 3 were investigated in CH2Cl2 solution by cyclic voltammetry (CV) in a three-electrode electrochemical cell with Bu4NClO4 (0.1 M) as electrolyte, and platinum, Ag/AgCl as the working electrode, counter electrode, and reference electrode, respectively; and using ferrocene (Fc/Fc+) as the internal standard with a scan rate of 100 mV s−1 at room temperature.As shown in Fig. 9, compound 3b exhibits a quasi-reversible oxidation processing the positive potential region with a oxidation process around 1.0 V (vs. Ag/AgCl), and compounds 3c, 3e and 3f exhibit two reversible or quasi-reversible oxidation processes, respectively. On the basis of the absorption spectra and the CV, the corresponding HOMO and LUMO energy levels were confirmed and the results are displayed in Table 2 and in the ESI (Table S3†). The HOMO values were calculated from the oxidation potential by the empirical formulae HOMO = −(4.8 + Eonsetox), where Eox is the onset of the oxidation potential. The HOMO values were −5.44 eV for 3b, −5.06 eV for 3c, −5.35 eV for 3e and −5.36 eV for 3f. The LUMO levels were determined from the HOMO and energy gap. For 3b and 3c containing increasing numbers of 4-methoxyphenyl moieties in different positions on the pyrene, similar energy gaps (3.17 eV for 3b and 3.13 eV for 3c) with different HOMO and LUMO levels are achieved.
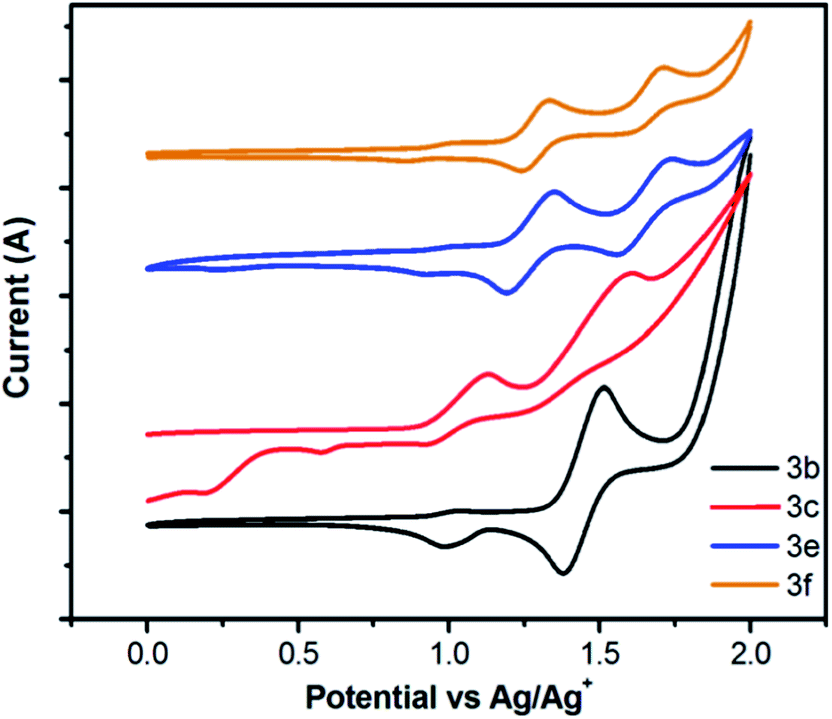 | ||
| Fig. 9 Cyclic voltammograms recorded for selected compounds 3. | ||
Additionally, through the same numbers of substituents located at different substituted positions in compounds 3e and 3f, that show similar HOMO (−5.36 eV for 3e and −5.37 eV for 3f, respectively) and LUMO values (−2.33 eV for 3e and −2.34 eV for 3f, respectively). From both Fig. 9 and Table S2,† it can be seen that the oxidation potentials are shifted to more positive values when increasing the number of substituents. The half-wave potentials for compounds 3b (2 substituents), 3c (3 substituents), 3e (4 substituents) and 3f (4 substituents) are 1.45 eV, 1.51 eV, 1.64 eV and 1.67 eV, respectively. Furthermore, the reduction potentials were dependent on substituent position, and a substituent at the active sites, namely the 1,3,6,8-positions, would lower the LUMO level, whereas, at the K-region of 4,5,9,10-position contributes to improve LUMO level.
Conclusions
In summary, the bromination mechanism of pyrene was explored by experimental methods. Clear evidence was observed for the formation of mono- to tetrakis(4-methoxyphenyl)-substituted pyrenes (3), which were synthesized by Suzuki–Miyaura cross-coupling reaction of the corresponding bromopyrenes with 4-methoxyphenyl boronic acid, and characterized by single-crystal X-ray diffraction, 1H/13C NMR spectra, mass spectrometry as well as elemental analysis. These results supported our conclusions on the bromination mechanism, namely that it was possible to regioselectively generate the mono- to tetrabromopyrenes from the 2-tert-butylpyrene (1) via a stepwise electrophilic substitution using an FeBr3-catalyzed rearrangement. Otherwise, the spectroscopic data, DFT calculations and electron chemistry results of the aryl-functionalized pyrenes indicate that the HOMOs and LUMOs level can be tuned by both the degree of substitution and the substituent position; the numbers of substituent moieties contribute to a higher oxidation potential and the reduction potential was attributed to the substituent position. The series of new molecular materials combines excellent optical features with reasonable thermal stabilities, making such molecules potential candidates in optoelectronic applications such as OLED-like devices. Further investigations on their usefulness in organic electroluminescent devices are in progress in our laboratory.Experimental section
Materials
Unless otherwise stated, all other reagents used were purchased from commercial sources and used without further purification. The preparation of 2-tert-butylpyrene (1)32 was reported previously.All melting points (Yanagimoto MP-S1) are uncorrected. 1H/13C NMR spectra (300 MHz) were recorded on a Nippon Denshi JEOL FT-300 NMR spectrometer. IR spectra were measured for samples as KBr pellets in a Nippon Denshi JIR-AQ2OM spectrophotometer. Mass spectra were obtained with a Nippon Denshi JMS-HX110A Ultrahigh Performance Mass Spectrometer at 75 eV using a direct-inlet system. Elemental analyses were performed by Yanaco MT-5. UV/vis spectra were obtained with a Perkin-Elmer Lambda 19 UV/vis/NIR spectrometer in various organic solvents. Fluorescence spectroscopic studies were performed in various organic solvents in a semimicro fluorescence cell (Hellma®, 104F-QS, 10 × 4 mm, 1400 μL) with a Varian Cary Eclipse spectrophotometer. Fluorescence quantum yields were measured using absolute methods. Thermogravimetric analysis (TGA) was undertaken using a SEIKO EXSTAR 6000 TG/DTA 6200 unit under a nitrogen atmosphere at a heating rate of 10 °C min−1. Differential scanning calorimeter (DSC) was performed using a Perkin-Elmer Diamond DSC Pyris instrument under a nitrogen atmosphere at a heating rate of 10 °C min−1. Photoluminescence spectra were obtained using a FluroMax-2 (Jobin-Yvon-Spex) luminescence spectrometer. Electrochemical properties of HOMO and LUMO energy levels were determined by a Electrochemical Analyzer. The quantum chemistry calculation was performed using the Gaussian 03W (B3LYP/6-31G* basis set) software package.33
Run 1: Lewis acid-catalysed bromination of 2-tert-butylpyrene. A mixture of 2-tert-butylpyrene (1) (0.26 g, 1 mmol) and iron powder (0.56 g, 10 mmol) were combined in CH2Cl2 (10 mL) at 0 °C with stirring for 30 min. A solution of Br2 (0.051 mL, 1 mmol) in CH2Cl2 (10 mL) was slowly added drop-wise with vigorous stirring. After this addition, the reaction mixture was continuously stirred for 5 h at 28 °C. The mixture was quenched with Na2S2O3 (10%) and extracted with CH2Cl2 (20 mL × 2). The combined organic extracts were washed with water and brine and evaporated. The crude product was a brown colour, and the residue was recrystallized from a mixed solution of toluene–hexane (1
:2) to give pure 1-bromo-7-tert-butylpyrene (2a) in 83% yield (290 mg) as a white powder.
Run 2. A solution of benzyltrimethylammonium tribromide (BMTABr3) (7.57 g, 19.4 mmol) in dry CH2Cl2 (50 mL) was slowly added to a solution of 2-tert-butylpyrene (1) (5 g, 19.4 mmol) in dry CH2Cl2 (150 mL) at 0 °C under a nitrogen atmosphere. The resulting mixture was allowed to slowly warm up to 28 °C and stirred overnight. The reaction mixture was quenched with Na2S2O3 and extracted with CH2Cl2 (2 × 50 mL). The combined organic extracts were dried with anhydrous MgSO4 and evaporated. The residue was crystallized from hexane to give pure 1-bromo-7-tert-butylpyrene (2a) in 84% yield (5.5 g) as white crystals. The 1H NMR spectrum completely agreed with the reported values.19b 1H NMR (300 MHz, CDCl3): δ (TMS, ppm) 1.59 (s, 9H, tBu), 7.99 (d, J = 8.2 Hz, 1H), 8.01 (d, J = 8.79 Hz, 1H), 8.08 (d, J = 9.0 Hz, 1H), 8.17 (d, J = 10 Hz, 1H), 8.20 (d, J = 8.22 Hz, 1H), 8.27–8.27 (m, 2H), 8.40 (d, J = 9.0 Hz, 1H). FAB-HRMS: m/z calcd for C20H17Br 337.3; found 335.9 [M+]. Anal. calcd for C20H17Br: C, 71.23; H, 5.08; found: C, 72.25; H, 6.33%.
Run 3. A mixture of 2-tert-butylpyrene (1) (0.26 g, 1 mmol) and iron powder (0.56 g, 10 mmol) were added in CH2Cl2 (10 mL) at 0 °C with stirring for 30 min. A solution of Br2 (0.105 mL, 2 mmol) in CH2Cl2 (10 mL) was slowly added drop-wise with vigorous stirring. After this addition, the reaction mixture was continuously stirred for 5 h at 28 °C. The mixture was quenched with Na2S2O3 (10%) and extracted with CH2Cl2 (20 mL × 2). The combined organic extracts were washed with water and brine and evaporated. The crude product was black in colour. The residue was washed with hot hexane and filtered to afford 1,3-dibromo-7-tert-butylpyrene (2b) 146 mg (35%). The solution was evaporated and recrystallized from a mixed solution of CH2Cl2–hexane (1
:2) to give pure 1-bromo-7-tert-butylpyrene (2a) in 50% yield (170 mg) as white powder.
Run 4. To a mixture of 2-tert-butylpyrene (1) (2.58 g, 10 mmol) in CH2Cl2 (30 mL) was added drop-wise a solution of BTMABr3 (benzyltrimethylammonium tribromide) (13.7 g, 35 mmol) in CH2Cl2 (20 mL) at 0 °C for 1 h under an argon atmosphere. The resulting mixture was allowed to slowly warm up to 28 °C and stirred overnight. The reaction mixture was poured into ice-water (60 mL) and neutralized with an aqueous 10% Na2S2O3 solution. The solution was extracted with CH2Cl2 (2 × 50 mL). The organic layer was washed with water (2 × 20 mL) and saturated brine (20 mL), and then the solution was dried (MgSO4) and condensed under reduced pressure. The crude compound was washed with hot hexane to afford pure 1,3-dibromo-7-tert-butylpyrene (2b) in 76% yield (3.02 g) as a colourless solid. Recrystallization from hexane gave 2b as a gray solid, Mp: 199.5–201.2 °C. The 1H NMR spectrum agreed with the reported values.19a 1H NMR (300 MHz, CDCl3): δ (TMS, ppm) 1.59 (s, 9H, tBu), 8.15 (d, J = 9.2 Hz, 2H, pyrene-H), 8.30 (s, 1H, pyrene-H), 8.47 (d, J = 9.2 Hz, 2H, pyrene-H), 8.47 (s, 1H, pyrene-H). FAB-HRMS: m/z calcd for C20H16Br2 416.2; found 417.4 [M+]. Anal. calcd for C20H16B2: C, 55.72; H, 3.88; found: C, 55.05; H, 4.56%.
Run 5: Synthesis of 1,3,6-tribromo-7-tert-butylpyrene (2c)22. 2-tert-Butylpyrene (1) (1.3 g, 5.03 mmol) was dissolved in CH2Cl2 (20 mL) and stirred for 30 min. at 25 °C. To this solution was added Br2 (1 mL, 19.4 mmol) and vigorously stirred for 24 h. The reaction mixture was poured into ice-water (50 mL) and neutralized with an aqueous 10% Na2S2O3 solution, the mixture was extracted with CH2Cl2 (2 × 50 mL), and the organic layer was washed with brine and evaporated. The residue was recrystallized from a mixed solution of CH2Cl2–hexane (2
:1) to afford 1,3,6-tribromo-7-tert-butylpyrene(2c) in 65% yield (1.6 g). Mp: 276–277 °C. The 1H NMR spectrum completely agreed with the reported values.22 1H NMR (300 MHz, CDCl3): δ (TMS, ppm) 1.81 (s, 9H, tBu), 8.08 (d, J = 9.2 Hz, 1H, pyrene-H), 8.10 (s, 1H, pyrene-H), 8.34 (d, J = 8.58 Hz, 1H, pyrene-H), 8.39 (d, J = 9.7 Hz, 1H, pyrene-H), 8.46 (s, 1H, pyrene-H), 8.85 (d, J = 9.7 Hz, 1H, pyrene-H). FAB-HRMS: m/z calcd for C20H15Br3 495.05; found 494.0 [M+]. Anal. calcd for C20H15Br3: C, 48.52; H, 3.05; found: C, 49.32; H, 4.55%.
Run 6: Lewis acid-catalyzed bromination of 2-tert-butylpyrene. A mixture of 2-tert-butylpyrene (1) (0.26 g, 1 mmol) and iron powder (0.56 g, 10 mmol) were added to CH2Cl2 (10 mL) at 0 °C with stirring for 30 min. A solution of Br2 (0.153 mL, 3 mmol) in CH2Cl2 (10 mL) was slowly added drop-wise with vigorous stirring. After this addition, the reaction mixture was continuously stirred for 5 h at 28 °C. The mixture was quenched with Na2S2O3 (10%) and extracted with CH2Cl2 (2 × 20 mL). The combined organic extracts were washed with water and brine and evaporated. The crude product was washed with hot hexane to afford a mixture of white compounds 2d and 2f (430 mg); the mixture is difficult to separate by common chromatography. The yield was evaluated by 1H NMR spectral analysis (25% for 2d, 50% for 2f). The product cannot further been separated by High-speed Counter-current Chromatography (HSCCC) and was used as a mixture for the Suzuki coupling reactions. 1H NMR (300 MHz, CDCl3) for 2d: δ (TMS, ppm) 1.64 (s, 9H, tBu), 8.79 (s, 2H, pyrene-H), 8.86 (d, J = 9.8 Hz, 1H, pyrene-H), 8.89 (d, J = 8.86 Hz, 1H, pyrene-H), 8.90 (s, 1H, pyrene-H), 8.92 (s, 1H, pyrene-H).
Run 7: Synthesis of 1,3,5,9-tetrabromo-7-tert-butylpyrene (2f). A mixture of 2-tert-butylpyrene (0.512 g, 2 mmol) and iron powder (0.56 g, 10 mmol) were added to CH2Cl2 (10 mL) at 0 °C with stirring for 15 min. A solution of Br2 (0.61 mL, 12 mmol) in CH2Cl2 (15 mmol) was slowly added drop-wise with vigorous stirring. After this addition, the reaction mixture was continuously stirred for 4 h at 28 °C. The mixture was quenched with Na2S2O3 (10%) and extracted with CH2Cl2 (2 × 50 mL). The combined organic extracts were washed with water and brine and evaporated. The crude product was gray in color. The crude product was insoluble in common organic solvents such as benzene, hexane, methanol etc. and slightly dissolved in CH2Cl2 or CHCl3. The residue was dissolved in hot CHCl3 and filtered, and the product was crystallized from CHCl3 to give pure 1,3,5,9-tetrabromo-7-tert-butylpyrene (2f) in 84% yield (978 mg) as a white powder. Mp: 303.4–305.0 °C. IR (KBr): νmax (cm−1) = 2962, 2365, 1579, 1523, 1461, 1425, 1392, 1363, 1267, 1195, 1132, 1027, 1012, 941, 877, 809, 655, 474. 1H NMR (300 MHz, CDCl3): δ (TMS, ppm) 1.65 (s, 9H, tBu), 8.47 (s, 1H, pyrene-H2), 8.71 (s, 2H, pyrene-H), 8.79 (s, 2H, pyrene-H). Due to the poor solubility in organic solvents it was not further characterized by 13C NMR spectroscopy. FAB-HRMS: m/z calcd for C20H14Br4 573.78; found 573.62 [M+]. Anal. calcd for C20H14Br4: C, 41.85; H, 2.46; found: C, 42.05; H, 2.53%.
Run 8: Lewis acid-catalysed bromination of 1,3,6-tribromo-7-tert-butylpyrene (2c). A mixture of 1,3,6-tribromo-7-tert-butylpyrene (2c) (200 mg, 0.4 mmol) and iron powder (100 mg, 1.8 mmol) were added to CH2Cl2 (15 mL) at 0 °C with stirring for 15 min. A solution of Br2 (0.052 mL, 1.01 mmol) in CH2Cl2 (5 mL) was slowly added dropwise with vigorous stirring. After this addition, the reaction mixture was continuously stirred for 4 h at 28 °C. The mixture was quenched with Na2S2O3 (10%) and extracted with CH2Cl2 (2 × 20 mL). The combined organic extracts were washed by water and brine and evaporated. The crude product was dried and identified by 1H NMR spectroscopy. The yield was evaluated by 1H NMR at 70% for 2e, 30% for 2f. The product was not further separated and used as the mixture for the Suzuki coupling reaction. The greenish-yellow crude product was insoluble in common organic solvents such as benzene, hexane and methanol, and slightly soluble in CH2Cl2 or CHCl3. So the crude product was recrystallized from CHCl3 to afford a small amount of 2e for 1H NMR spectra analysis, 1H NMR (300 MHz, CDCl3): δ (TMS, ppm) 1.83 (s, 9H, tBu), 8.46 (d, J = 9.5 Hz, 1H, pyrene-H), 8.51 (s, 1H, pyrene-H), 8.76 (s, 1H, pyrene-H), 8.85 (s, 1H, pyrene-H), 8.91 (d, J = 9.9 Hz, 1H, pyrene-H).
Run 9: Lewis acid-catalysed bromination of 2e/2f. A mixture of 2e/2f (100 mg, 0.17 mmol) and iron powder (50 mg, 0.9 mmol) were added to CH2Cl2 (15 mL) at 0 °C with stirring for 15 min. A solution of Br2 (0.025 mL, 0.49 mmol) in CH2Cl2 (5 mL) was slowly added drop-wise with vigorous stirring. After this addition, the reaction mixture was continuously stirred for 8 h at 28 °C. The mixture was quenched with Na2S2O3 (10%) and extracted with CH2Cl2 (2 × 20 mL). The combined organic extracts were washed with water and brine and evaporated. The crude product (85 mg) was dried and identified by 1H NMR spectroscopy. The yield was evaluated by 1H NMR spectral analysis (25% for 2e, 75% for 2f).
:1) to give 3a as a white solid. Recrystallization from (CH2Cl2–hexane, 1:1) gave 7-tert-butyl-1-(4-methoxyphenyl) pyrene (3a) in 54% yield (235 mg) as colourless crystals. Mp: 141.2–143.5 °C. IR (KBr): νmax (cm−1) = 2952, 1604, 1520, 1496, 1437, 1246, 1172, 1032, 878, 849, 826, 729, 681, 567, 526, 489. 1H NMR (300 MHz, CDCl3): δ (TMS, ppm) 1.58 (s, 9H, tBu), 3.93 (s, 3H, OMe), 7.10 (d, J = 8.7 Hz, 2H, Ar–H), 7.56 (d, J = 8.7 Hz, 2H, Ar–H), 7.92 (d, J = 7.8 Hz, 1H, pyrene-H), 7.99 (d, J = 9 Hz, 1H, pyrene-H), 8.05 (s, 2H, pyrene-H), 8.15 (d, J = 1.8 Hz, 1H, pyrene-H), 8.18 (d, J = 2.4 Hz, 1H, pyrene-H), 8.22 (d, J = 1.8 Hz, 1H, pyrene-H). 13C NMR (100 MHz, CDCl3): δ (TMS, ppm) 158.9, 149.1, 137.2, 133.7, 131.6, 131.3, 130.8, 130.2, 128.4, 127.5, 127.4, 127.29, 127.26, 125.2, 124.9, 124.4, 123.2, 122.3, 122.0, 113.8, 55.4, 35.2, 31.9. FAB-HRMS: m/z calcd for C27H24O 364.18; found 364.25 [M+]. Anal. calcd for C27H24O: C, 88.97; H, 6.64; found: C, 88.68; H, 6.52%.:1) (113 mg, 69%). Mp: 167 °C. IR (KBr): νmax (cm−1) = 2958, 1610, 1512, 1498, 1456, 1396, 1363, 1286, 1246, 1174, 1039, 877, 835, 727, 660, 607, 580, 553, 418. 1H NMR (300 MHz, CDCl3): δ (TMS, ppm) 1.58 (s, 9H, pyrene-tBu), 3.93 (s, 6H, OMe), 7.10 (d, J = 8.8 Hz, 4H, Ar–H), 7.60 (d, J = 8.6 Hz, 4H, Ar–H), 7.91 (s, 1H, pyrene-H), 8.00 (d, J = 9.4 Hz, 2H, pyrene-H), 8.18 (d, J = 9.33 Hz, 2H, pyrene-H), 8.19 (s, 2H, pyrene-H). 13C NMR (75 MHz, CDCl3): δ (TMS, ppm) 159.0, 149.1, 136.8, 133.5, 131.7, 131.2, 129.1, 127.6127.4, 125.4, 125.2, 123.5, 122.0, 113.8, 55.4, 35.2, 31.9. FAB-HRMS: m/z calcd for C34H30O2 470.22; found 470.2 [M+]. Anal. calcd for C34H30O2: C, 86.77; H, 6.43; found: C, 86.53; H, 6.41%.:1) (155 mg, 67%). Mp: 249 °C. 1H NMR (300 MHz, CDCl3): δ (TMS, ppm) 1.41 (s, 9H, pyrene-tBu), 3.86 (s, 3H, OMe), 3.91 (s, 6H, OMe), 7.00 (d, J = 8.0 Hz, 4H, Ar–H), 7.08 (d, J = 8.4 Hz, 2H, Ar–H), 7.24–7.36 (m, 1H, pyrene-H, 2H, Ar–H), 7.51 (d, J = 8.4 Hz, 2H, Ar–H), 7.58 (d, J = 8.4 Hz, 2H, Ar–H), 7.89 (s, 1H, pyrene-H), 7.97 (d, J = 9.6 Hz, 1H, pyrene-H), 8.00 (d, J = 9.6 Hz, 1H, pyrene-H), 8.18 (d, J = 9.2 Hz, 2H, pyrene-H), 8.37 (s, 1H, pyrene-H). 13C NMR (100 MHz, CDCl3): δ (TMS, ppm) 158.9, 158.88, 158.7, 146.3, 136.8, 136.6, 136.58, 134.1, 133.6, 133.4, 133.1, 132.1, 131.7, 131.6, 130.5, 129.2, 127.9, 127.5, 127.1, 126.0, 125.1, 124.7, 123.4, 123.4, 113.8, 113.7, 112.8, 55.38, 55.36, 55.29, 37.2, 33.1. FAB-HRMS: m/z calcd For C41H36O3 576.27; found 576.34 [M+]. Anal. calcd for C41H36O3: C, 85.39; H, 6.29; found: C, 85.53; H, 6.21%.:1) to give a mixture of 7-tert-butyl-1,3,5-tris-(4-methoxyphenyl)pyrene 3d and 7-tert-butyl-1,3,5,9-tetrakis(4-methoxyphenyl)-pyrene 3f (20 mg) as yellow prisms. The mixture was recrystallized from toluene solution to afford a few crystals of 3d; the mixture was difficult to purify by HSCCC. Further detailed information (such 13C NMR spectroscopy, MS, elemental analysis) of 3d could not be obtained. Increasing the polarity of the eluant with CH2Cl2 gave only 7-tert-butyl-1,3,5,9-tetrakis(4-methoxyphenyl)pyrene 3f (15 mg) as a yellow solid. The 1H NMR spectrum of 3f agreed with the reported values.18 Mp: 407.5 °C. IR (KBr): νmax (cm−1) = 2954, 1610, 1510, 1461, 1442, 1367, 1288, 1246, 1174, 1107, 1036, 831, 586, 543. 1H NMR (300 MHz, CDCl3): δ (TMS, ppm) 1.38 (s, 9H, tBu), 3.90 (s, 6H, OMe), 3.92 (s, 6H, OMe), 7.05 (d, J = 1.6 Hz, 4H, Ar–H), 7.08 (d, J = 3.1 Hz, 4H, Ar–H), 7.57 (d, J = 8.8 Hz, 4H, Ar–H), 7.60 (d, J = 4.4 Hz, 4H, Ar–H), 7.92 (s, 1H, pyrene-H), 8.12 (s, 2H, pyrene-H), 8.30 (s, 2H, pyrene-H). 13C NMR (75 MHz, CDCl3): δ (TMS, ppm) 159.0, 158.9, 148.5, 139.1, 136.7133.9, 133.7, 131.7, 131.2, 130.8, 129.7, 127.3, 125.6, 121.2, 113.9113.8, 55.4, 31.7. FAB-HRMS: m/z calcd for C48H42O4 682.84; found 682.19 [M+]. Anal. calcd for C48H42O4: C, 84.43; H, 6.20; found: C, 84.33; H, 6.52%.:1) gave 3e in 42% yield (100 mg) as a yellow powder. Increasing the polarity of the eluant with CH2Cl2 afforded 7-tert-butyl-1,3,5,9-tetrakis(4-methoxy-phenyl)pyrene 3f in 17% yield (40 mg) as a yellow solid.:1). Mp: 240.0–241.2 °C. IR (KBr): νmax (cm−1) = 2954, 1607, 1507, 1462, 1440, 1288, 1246, 1177, 1105, 1037, 832, 808, 548, 526, 472. 1H NMR (300 MHz, CDCl3): δ (TMS, ppm) 1.30 (s, 9H, pyrene-tBu), 3.88 (s, 3H, OMe), 3.89 (s, 3H, OMe), 3.92 (s, 6H, OMe), 7.00 (m, 8H, Ar–H), 7.29 (d, J = 8.4 Hz, 2H, Ar–H), 7.35 (d, J = 9.5 Hz, 2H, pyrene-H), 7.54 (d, J = 8.61 Hz, 2H, 2H, Ar–H), 7.59 (d, J = 8.43 Hz, 2H, Ar–H), 7.60 (d, J = 8.61 Hz, 2H, Ar–H), 7.90 (s, 1H, pyrene-H), 7.97 (d, J = 9.69 Hz, 1H, pyrene-H), 8.12 (s, 1H, pyrene-H), 8.51 (s, 1H, pyrene-H). 13C NMR (100 MHz, CDCl3): δ (TMS, ppm) 159.0, 158.9, 158.7, 146.0, 138.9, 136.9, 136.6, 136.3, 134.2, 133.6, 133.5, 133.1, 132.2, 131.62, 131.59, 131.2, 129.7, 129.5, 127.7, 127.0, 126.1, 125.5, 124.6, 124.5, 123.8, 122.5, 113.9, 113.81, 113.77, 113.71, 112.9, 55.35, 55.29, 37.5, 33.0. FAB-HRMS: m/z calcd for C48H42O4 682.84; found 682.66 [M+]. Anal. calcd for C48H42O4: C, 84.43; H, 6.20; found: C, 81.90; H, 6.15%. The 1H NMR spectrum of 3f agreed with the reported values.18:1) gave 7-tert-butyl-1,3,5,9-tetrakis(4-methoxyphenyl)pyrene (3f) in 65% yield (154 mg) as a yellow solid.Crystallography for 3
Diffraction data were collected using a Rigaku R-Axis Rapid for 3a, or a Saturn724 for 3b. A Bruker SMART CCD was used for 3c, a SMART CCD for 3d, and APEX II CCD diffractometers in the home laboratory for 3c and 3f or at the Advanced Light Source (ALS) Station 11.3.1 for 3e.34 Data were corrected for absorption on the basis of symmetry equivalent and repeated data and Lp effects. Structural solution and full matrix least-squares refinement were performed with the SHELXS-97 and SHELXL-97 or SHELXL-2013 software packages, respectively.35 The structures were solved by direct methods and refined on F2 using all data. 36H atoms were constrained in a riding model. In 3c the methyl groups on the tBu group at C(17) was modeled with two sets of positions with major occupancy 63.4(12)%.†Acknowledgements
This work was performed under the Cooperative Research Program of “Network Joint Research Center for Materials and Devices (Institute for Materials Chemistry and Engineering, Kyushu University)”. We would like to thank the OTEC at Saga University and the International Collaborative Project Fund of Guizhou province at Guizhou University for financial support. We also would like to thank the EPSRC and the Royal Society of Chemistry (travel grants to CR) and The Scientific Research Common Program of Beijing Municipal Commission of Education for financial support. Thanks Dr Kai Chen (Nanjing University) for refining the X-ray diffraction data for 3a, 3b and 3d. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract no. DE-AC02-05CH11231.Notes and references
- (a) T. M. Figueira-Duarte and K. Müllen, Chem. Rev., 2011, 111, 7260–7314 CrossRef CAS PubMed; (b) J.-Y. Hu and T. Yamato, Org. Light Emitting Diode: Mater., Process Devices, 2011, 21–60 CAS; (c) P. C. Bevilacqua, R. Kierzek and K. A. Johnson, Science, 1992, 258, 1355–1358 CAS.
- A. G. Crawford, A. D. Dwyer, Z.-Q. Liu, A. Steffen, A. Beeby, L.-O. Pålsson, D. L. Tozer and T. B. Marder, J. Am. Chem. Soc., 2011, 133, 13349–13362 CrossRef CAS PubMed.
- D. N. Coventry, A. S. Batsanov, A. E. Goeta, J. A. K. Howard, T. B. Marder and R. N. Perutz, Chem. Commun., 2005, 2172–2174 RSC.
- A. G. Crawford, Z.-Q. Liu, I. A. I. Mkhalid, M.-H. Thibault, N. Schwarz, G. Alcaraz, A. Steffen, J. C. Collings, A. S. Batsanov, J. A. K. Howard and T. B. Marder, Chem.–Eur. J., 2012, 18, 5022–5035 CrossRef CAS PubMed.
- S.-T. Lin, Y.-F. Jih and P. P. Fu, J. Org. Chem., 1996, 61, 5271–5273 CrossRef CAS.
- H. Vollmann, M. Becker, M. Correl and H. Streeck, Justus Liebigs Ann. Chem., 1937, 531, 1–159 CrossRef CAS.
- T. Yamato, A. Miyazawa and M. Tashiro, J. Chem. Soc., Perkin Trans. 1, 1993, 3127–3137 RSC.
- T. Yamato, M. Fujimoto, A. Miyazawa and K. Matsuo, J. Chem. Soc., Perkin Trans. 1, 1997, 1201–1207 RSC.
- J. Hu, D. Zhang and F. W. Harris, J. Org. Chem., 2005, 70, 707–708 CrossRef CAS PubMed.
- (a) X. Feng, F. Iwanaga, J.-Y. Hu, H. Tomiyasu, M. Nakano, C. Redshaw, M. R. J. Elsegood and T. Yamato, Org. Lett., 2013, 15, 3594–3597 CrossRef CAS PubMed; (b) B.-X. Gao, M. Wang, Y.-X. Cheng, L.-X. Wang, X.-B. Jing and F.-S. Wang, J. Am. Chem. Soc., 2008, 130, 8297–8306 CrossRef CAS PubMed; (c) N. Kulisic, S. Moreab and A. Mateo-Alonso, Chem. Commun., 2011, 47, 514–516 RSC; (d) A. Mateo-Alonso, Chem. Soc. Rev., 2014, 43, 6311–6324 RSC.
- J. Se. Kim and D. T. Quang, Chem. Rev., 2007, 107, 3780–3799 CrossRef CAS PubMed.
- L. Chouai, F.-Y. Wu, Y. Jang and R. P. Thummel, Eur. J. Inorg. Chem., 2003, 2774–2782 CrossRef CAS.
- G. Venkataramana and S. Sankararaman, Org. Lett., 2006, 8, 2739–2742 CrossRef CAS PubMed.
- J. N. Moorthy, P. Natarajan, P. Venkatakrishnan, D.-F. Huang and T. Chow, Org. Lett., 2007, 9, 5215–5218 CrossRef CAS PubMed.
- Y. Niko, S. Kawauchi, S. Otsu, K. Tokumaru and G.-I. Konishi, J. Org. Chem., 2013, 78, 3196–3207 CrossRef CAS PubMed.
- X.-L. Ni, X. Zeng, C. Redshaw and T. Yamato, J. Org. Chem., 2011, 76, 5696–5702 CrossRef CAS PubMed.
- (a) J.-Y. Hu, X.-L. Ni, X. Feng, M. Era, M. R. J. Elsegood, S. J. Teatd and T. Yamato, Org. Biomol. Chem., 2012, 10, 2255–2262 RSC; (b) J.-Y. Hu, M. Era, M. R. J. Elsegood and T. Yamato, Eur. J. Org. Chem., 2010, 72–79 CrossRef.
- X. Feng, J.-Y. Hu, F. Iwanaga, N. Seto, C. Redshaw, M. R. J. Elsegood and T. Yamato, Org. Lett., 2013, 15, 1318–1321 CrossRef CAS PubMed.
- (a) X. Feng, J.-Y. Hu, L. Yi, N. Seto, Z. Tao, C. Redshaw, M. R. J. Elsegood and T. Yamato, Chem.–Asian J., 2012, 7, 2854–2863 CrossRef CAS PubMed; (b) T. M. Figueira-Duarte, S. C. Simon, M. Wagner, S. I. Druzhinin, K. A. Zachariasse and K. Müllen, Angew. Chem., Int. Ed., 2008, 47, 10175–10178 CrossRef CAS PubMed.
- L. M. Sayre and F. R. Jensen, J. Am. Chem. Soc., 1979, 101, 6001–6008 CrossRef CAS.
- (a) A. Burritt, J. M. Coxon and P. J. Steel, J. Org. Chem., 1996, 61, 4328–4335 CrossRef CAS PubMed; (b) X. Gao, W. Qiu, X. Yang, Y. Liu, Y. Wang, H. Zhang, T. Qi, Y. Liu, K. Lu, C. Du, Z. Shuai, G. Yu and D.-B. Zhu, Org. Lett., 2007, 9, 3917–3920 CrossRef CAS PubMed.
- J. Inoue, K. Fukui, T. Kubo, S. Nakazawa, K. Sato, D. Shiomi, Y. Morita, K. Yamamoto, T. Takui and K. Nakasuji, J. Am. Chem. Soc., 2001, 123, 12702–12703 CrossRef CAS PubMed.
- W. E. Moffitt and C. A. Coulson, Proc. Phys. Soc., London, 1948, 60, 309–315 CrossRef CAS.
- A. S. Batsanov, J. A. K. Howard, D. Albesa-Jové, J. C. Collings, Z. Q. Liu, I. A. I. Mkhalid, M.-H. Thibault and T. B. Marder, Cryst. Growth Des., 2012, 12, 2794–2802 CAS.
- V. de. Halleux, J.-P. Callbert, P. Brocorens, J. Cornil, J.-P. Declercq, J.-L. Brédas and Y. Geerts, Adv. Funct. Mater., 2004, 14, 649–659 CrossRef.
- K. R. J. Thomas, J. T. Lin, Y.-T. Tao and C.-H. Chuen, Chem. Mater., 2002, 14, 2796–2802 CrossRef CAS.
- (a) F. Polo, F. Rizzo, M. Veiga-Gutierrez, L. D. Cola and S. Quici, J. Am. Chem. Soc., 2012, 134, 15402–15409 CrossRef CAS PubMed; (b) S. Kato, S. Shimizu, H. Taguchi, A. Kobayashi, S. Tobita and Y. Nakamura, J. Org. Chem., 2012, 77, 3222–3232 CrossRef CAS PubMed.
- P. Tyagi, A. Venkateswararao and K. R. J. Thomas, J. Org. Chem., 2011, 76, 4571–4581 CrossRef CAS PubMed.
- Y. Li, J. Ding, M. Day, Y. Tao, J. Lu and M. D'iorio, Chem. Mater., 2004, 16, 2165–2173 CrossRef CAS.
- C.-G. Wang, S.-Y. Chen, K. Wang, S.-S. Zhao, J.-Y. Zhang and Y. Wang, J. Phys. Chem. C, 2012, 116, 17796–17806 CAS.
- Z.-Q. Liu, Y.-Y. Wang, Y. Chen, J. Liu, Q. Fang, C. Kleeberg and T. B. Marder, J. Org. Chem., 2012, 77, 7124–7128 CrossRef CAS PubMed.
- Y. Miura, E. Yamano, A. Tanaka and J. Yamauchi, J. Org. Chem., 1994, 59, 3294–3300 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery Jr, T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. G. Stefanov, A. Liu, P. Liashenko, I. Piskorz, R. L. Komaromi, D. J. Martin, T. Fox, M. A. Keith, C. Y. Al-Laham, A. Peng, M. Nanayakkara, P. Challacombe, M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian 03, revision C.02, Gaussian, Inc., Wallingford CT, 2004 Search PubMed.
- APEX II and SAINT, Software for diffractometers, Bruker AXS Inc., Madison, WI, USA, 2006 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A, 2008, 64, 112–120 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR and 13C NMR spectra of 2 and 3, IR spectra, X-ray data for 3 (CIF) and optimized structure. CCDC 965757, 879771, 965758, 965759, 965760 and 917257. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ra12216j |
| This journal is © The Royal Society of Chemistry 2015 |