
Effect of alkyl substituents in BODIPYs: a comparative DFT computational investigation†
Sanjoy Mukherjee and
Pakkirisamy Thilagar*
Department of Inorganic and Physical Chemistry, Indian Institute of Science, Bangalore-560012, India. E-mail: sanjoymkj@ipc.iisc.erent.in; thilagar@ipc.iisc.ernet.in; Tel: +91-80-2293-3353
First published on 1st December 2014
Abstract
Random changes in the alkyl substitution patterns of fluorescent dyes, e.g. BODIPYs, are often accompanied by significant changes in their photophysical properties. To understand such alterations in properties in closely related molecular systems, a comparative DFT (density functional theory) computational investigation was performed in order to comprehend the effects of alkyl substitution in controlling the structural and electronic nature of BODIPY dyes. In this context, a systematic strategy was utilized, considering all possible outcomes of constitutionally-isomeric molecules to understand the alkyl groups’ effects on the BODIPY molecules. Four different computational methods {i.e. B3LYP/6-31G(d); B3LYP/6-311++G(d,p); wb97xd/6-311++G(d,p) and mpw1pw91/6-311++G(d,p)} were employed to rationalize the agreement of the trends associated with the molecular properties. In line with experimental observations, it was found that alkyl substituents in BODIPY dyes situated at 3/5-positions effectively participate in stabilization as well as planarization of such molecules. Screening of all the possible isomeric molecular systems was used to understand the individual properties and overall effects of the typical alkyl substituents in controlling several basic properties of such BODIPY molecules.
Introduction
The recent progress of luminescent materials would be largely incomplete if one does not take into account BODIPYs (boron-dipyrromethenes).1 BODIPYs are a considerably large class of tetracoordinate boron-containing dyes, where the dipyrrin-type ligands act as the chelating groups towards the boron atom. With their sharp and tunable absorption and emission profiles, coupled with their photo-stability and frequently observed high quantum yields, BODIPYs have found paths towards almost all modern applications. BODIPYs find applications in biological live-cell imaging,2 fluorescence recognition,3 light-harvesting systems,4 photo-catalysis,5 etc. Although the progress of applications of BODIPY-based compounds have been remarkable, the origins of their excellent properties are yet not well-understood.The prediction of the electronic properties of BODIPY compounds using available computational tools and theoretical models mostly deviate considerably from the actual experimentally observed properties.6 Despite the limitations, the usage of computational methods, such as DFT, etc., has found an important role in understanding the chemistry of boron-based dyes.7 The comparative nature of closely related molecules is better understood from regular computational results rather than the exact natures of the individual compounds. In recent times, a number of computational efforts have been implemented to gain a comparative insight into the photophysics of BODIPY dye analogues.6,8 However, prior to this report, there has been no substantial work on the effect of alkyl substituents in controlling the nature of these dyes. It is notable that due to the synthetic convenience, and to control physical properties e.g. solubility, alkyl substituents are often preferred on the pyrollic moieties of BODIPYs. However, it has been observed that, in many cases, such alkyl groups significantly alter the photophysical properties of BODIPYs.9,10 In this work, a closer theoretical perspective of such observations is explored. A comparative understanding of the effect of alkyl groups in their control over the total energy, ring-planarity and FMO energies of BODIPY dyes are discussed.
The molecular structure of the BODIPY core closely resembles the geometry of indacenes. As shown in Fig. 1, a close look at the {B3LYP/6-31G(d) optimized} molecular structure (see the ESI†) would suggest that the substituents at the meso-position would experience comparatively less steric interaction with neighboring groups, whereas the same is expected to be comparatively greater for the 1, 2, 6 and 7-positioned substituents. Due to the presence of a borate moiety, the substituents at the 3 and 5 positions are expected to experience the highest extent of steric interaction with the neighboring environment. If the interactions of such substituents are solely governed by steric interactions, 3/5-alkyl substituted BODIPYs can be expected to be energetically less stable or puckered compared to their constitutional isomers. However, the experimental observations found in previous reports, as well as the computational results found in this work, provide completely opposite results.
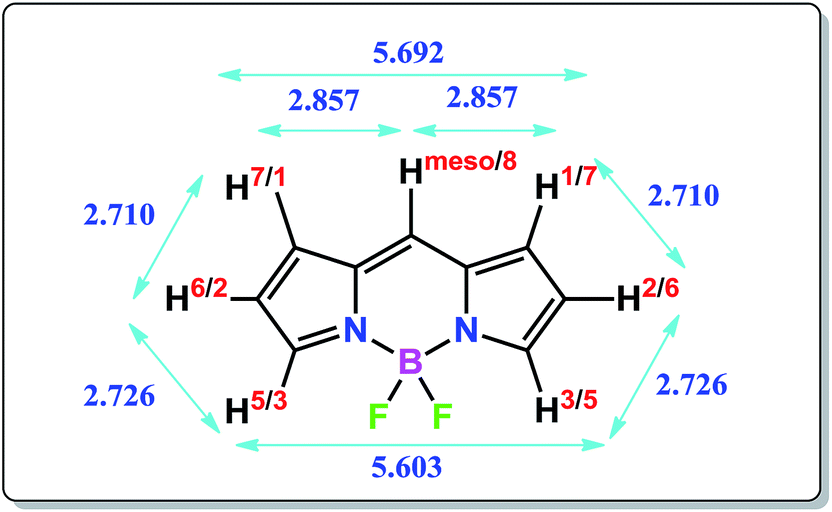 | ||
| Fig. 1 Molecular structure of BODIPY core showing atom numbering scheme and distances between neighboring H atoms in angstroms (B3LYP/6-31G(d) optimized structure). | ||
Experimental observations often encounter uncorrelated behaviors of different 3/5-substituted BODIPYs. Previous reports from Boens et al., and Cabrera and Tang et al.,9 followed by our recent investigations,10 have demonstrated that the alkyl substituents on BODIPY dyes can significantly alter their photophysical properties. As shown in Fig. 2, in the series of BODIPYs depicted in the scheme, the 3,5-dimethyl substituted compounds show significantly red-shifted absorption (∼10–15 nm) profiles compared to the BODIPYs with no substituents at all or with 1,3,5,7-tetramethyl substituents. As the first and third members of the last two series show the same absorption pattern based on the BODIPY core, the sudden alteration of the band gap of the 3,5-dimethyl substituted BODIPYs cannot be accounted for by considering only the π–π conjugation throughout the molecular backbone. Also, the 3,5-dimethyl substituted BODIPYs show higher quantum efficiencies than the BODIPYs with no methyl substituents. As observed, the 3,5-dimethyl substituents participate actively in somehow rigidifying the BODIPY system and also effectively diminishing the effective band-gap. Such small changes in electronic and structural properties result in considerably great effects on the photophysical properties of BODIPYs and other multichromophoric molecular conjugates.11 In order to understand the effect of alkyl substituents in controlling the properties of BODIPYs, a comparative computational study was performed, considering all possibilities of such substitution patterns.
 | ||
| Fig. 2 Comparison of optical properties of several series of structurally related BODIPY compounds differing only in the alkyl substitution on the BODIPY unit (ref. 9 and 10). | ||
Methodology
All the density functional theory (DFT) calculations were performed using standard computational methods and basis sets as incorporated in the Gaussian 09 software package.12 The most commonly used B3LYP functional, with 6-31G(d) basis sets for all the atoms, was used in this regard (Table S1-1, ESI†).13 Frequency tests of the optimized structures were performed to ascertain stationary points. Additionally, the calculations were performed using the B3LYP/6-311++G(d,p), wb97xd/6-311++G(d,p) [ref. 14] and mpw1pw91/6-311++G(d,p) [ref. 15] methods to confirm that the results obtained in the previous method were not dependent on the choice of either functional or basis-sets (see the ESI†). Time-dependent DFT (TD-DFT) 1st excited state geometry optimizations were performed using only the B3LYP/6-31G(d) methodology.In order to obtain a complete understanding of the effect of small alkyl groups on the nature of BODIPY dyes, systematic alterations of substituents were carried out. For this purpose, five hypothetical series of compounds were taken into account for the computational studies (Fig. 3). Series 1 consists of all possible structures with a single methyl substitution around the BODIPY core. As only four possibilities arise, models 1.01 to 1.04 are the constituents of this series. Similarly, series 3 considers single ethyl substitution, whereas series 5 refers to single tert-butyl substitution, which was taken into consideration for understanding the steric effects of the alkyl groups. On the other hand, series 2 consists of possibilities where two simultaneous methyl substitutions are present around the BODIPY core. In this case, twelve different possibilities arise, which provides a relatively large series for comparative understanding of the constitutional isomers. Similarly, series 4 consists of models that feature two simultaneous ethyl substitutions. In the complete picture, a total of 37 model systems were taken into account.
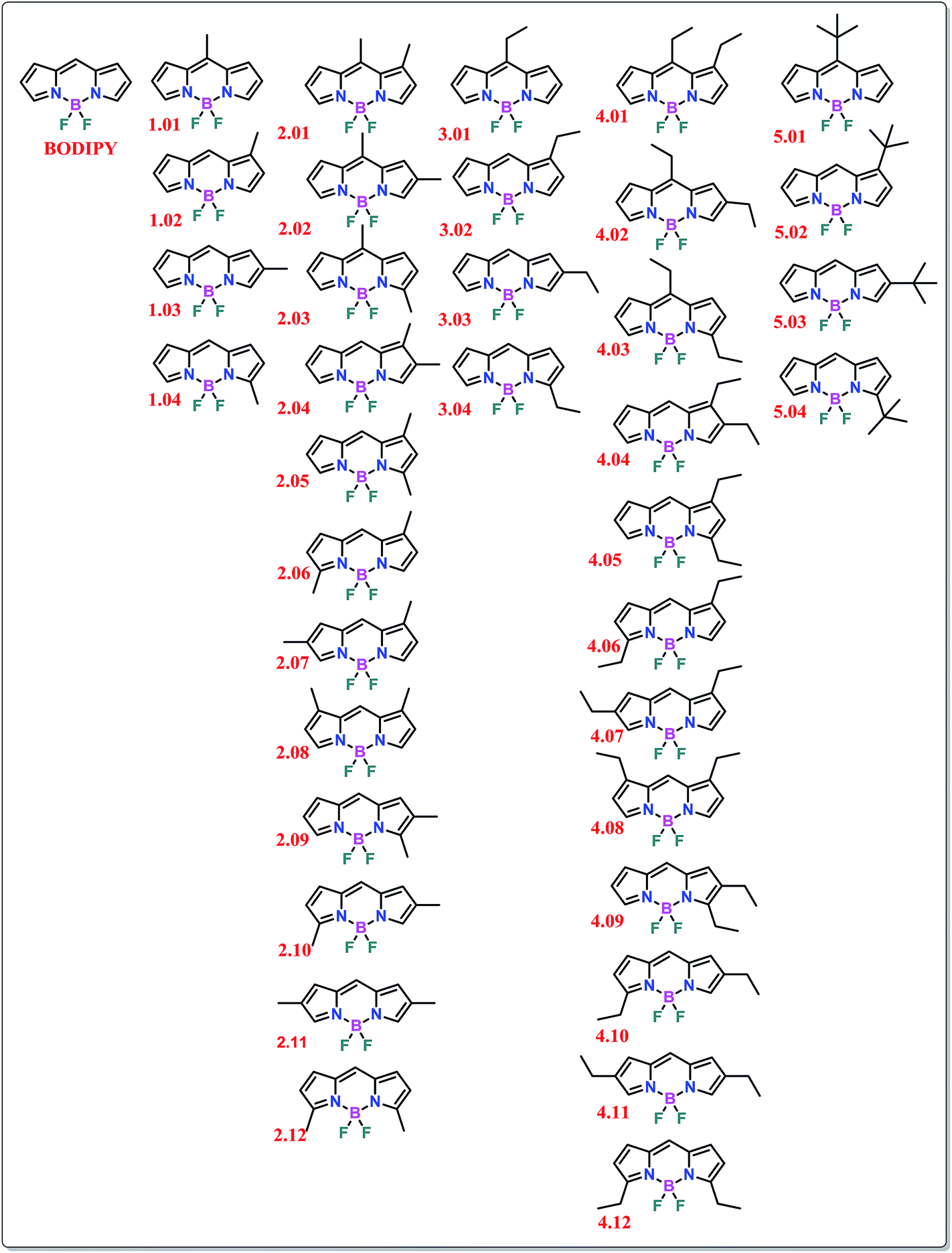 | ||
| Fig. 3 Structural formulae of the BODIPY compounds taken into consideration in the current study. | ||
Effect on relative stability
Although the individual members of a particular series are related to one another as constitutional isomers, their total energies, i.e. stabilities, differ greatly (see the ESI†). As shown in Fig. 1, the steric interactions between neighboring C1 and C8 substituents are expected to be less prominent than between the C3/C5 and BF2 centers. However, the computational results differed vastly from our general expectations. As shown in Fig. 4, in all the series 1–4, the most stable isomers are those with substituents situated at the 3 and/or 5 positions of the BODIPY moiety. The only exception is found in series 5, where the most stable isomer is 5.03. It is also noteworthy that in all the series 1–5, the highest energy isomers are those decorated with C8-substituents. The trends found in this regard are independent of the choice of functional or usage of either 6-31G(d) or 6-31++G(d,p) basis sets. As evident from the results obtained using B3LYP/6-31G(d), model system 1.04 is a ∼15.2 kJ mol−1 more stable isomer compared to 1.01. This is also followed in series 3, where 3.04 experiences a ∼17.0 kJ mol−1 stability increase compared to 3.01. The minute differences in the relative stabilization energies of the 3-substituted isomers (i.e. 1.04 and 2.04) compared to the meso-substituted isomers (i.e. 1.01 and 3.01) indicate that these effects are mostly not due to steric interactions (see the ESI†). | ||
| Fig. 4 Comparison of the relative total energies of the BODIPY isomers present in each series (series 1–5) as obtained from DFT B3LYP/6-31G(d) optimizations. | ||
The trends observed in series 2 and series 4 further support that the lower ends of the energy profiles in the series of constitutional isomers are mostly held by the 3-substituted members (i.e. 2.03, 2.05, 2.06, 2.09, 2.10 and 2.12 for series 2, and similarly for series 4) whereas the 3,5-disubsituted members (i.e. 2.12 and 4.12) are the most stable isomers, with stabilization energies (compared to 2.01 and 4.01, respectively) of ∼34.0 kJ mol−1 for series 2 and ∼43.2 kJ mol−1 for series 4. The trends in these two groups suggest that steric interactions between neighboring groups are most prominent for the meso-substituted compounds, whereas the effects are least prominent for the 3 (and/or 5) substituted isomers. Compounds 2.09 and 4.09 are relatively stable isomers compared to 2.11 and 4.11, which is an apparent anomaly if one considers only steric interactions playing a role in such energy differences. For instance, 4.09 contains two neighboring bulky ethyl units, whereas in 4.11 no such steric interactions are present except ethyl–hydrogen steric interactions. This is also evident from the observed trends in series 5, where 5.03 is the most stable isomer among 5.01–5.04. The steric demand for 2,6-subsituted BODIPYs (e.g. 4.11) should have been less than that for 2,3-substituted BODIPYs (e.g. 4.09). The explanations based on steric factors cannot completely justify the observations and the overall results indicate the active participation of the alkyl substituents in governing the overall features of the BODIPY systems.
Effect on FMOs
Further justifications of our interpretations follow from the tendencies observed in the FMO energies of the model BODIPY systems (see the ESI†). As shown in Fig. 5, in series 1 and 3 the relative energies of the HOMO orbitals increase gradually in an almost linear fashion going from the meso-substituted (1.01 or 3.01) to the 3-subsituted isomers (1.04 or 3.04, respectively). The overall increments observed in these cases are rather considerable (∼0.13 eV for series 1 and ∼0.23 eV for series 3); in complex molecular systems, such changes induced by mere alkyl substituents might have great effects on photophysical properties such as energy or electron transfer processes. Observations of the trends in the HOMO energies of series 2 and 4 make it evident that the trends are quite the opposite to those observed in the total energy of the molecules. In these cases, the peaks of the energy profiles are held by the 3 (and/or 5) substituted isomers, whereas the troughs are constituted of the 1-substituted isomers. As shown in Fig. 6, the interactions of the C–H bonding orbitals with the BODIPY-based orbitals are effectively involved in obtaining these trends. The meso-methyl substitution in compound 2.01 contributes by a rather low amount to the formation of the HOMO (compared to others in the same series). It is also qualitatively evident that the involvement of the C–H δ-bonds in the HOMO increases gradually from 2.02 to 2.04. In all cases, it is found that the C–H δ-bonding orbitals are in destructive interference with the BODIPY-centered orbitals and thus can only increase the HOMO energy. Consideration of this idea alone can clearly describe the complete trends as observed in series 1–4. For instance, in series 2, model 2.01 has the lowest HOMO energy. A linear increase is followed up to 2.03, where the 3-subsitution results in a comparatively higher HOMO energy. By a similar argument, model 2.12 has the highest HOMO energy resulting from the participation of the C–H δ-bonds of the 3 and 5 substituents. | ||
| Fig. 5 Comparison of the relative HOMO (left) and LUMO (right) energies of the BODIPY isomers represented in each series (series 1–4) as obtained from DFT B3LYP/6-31G(d) optimizations. | ||
 | ||
| Fig. 6 DFT B3LYP/6-31G(d)-obtained HOMOs of 1.01 to 1.04 (from left to right, respectively; isovalue = 0.02). | ||
Different observations are followed in case of the LUMOs of the model BODIPY systems. In series 1, the LUMO energies decrease gradually from 1.01 to 1.03, whereas the LUMO energy of 1.04 is highest in series 1. Similar observations are followed for series 3 and series 5. The observations can be explained well by considering the interactions observed in these LUMO orbitals. As shown in Fig. 7, from 1.01 to 1.03, the effective involvement of C–H δ-orbitals in the formation of the LUMOs gradually decreases, whereas it regains its efficiency in 1.04. It is also evident that the 2-subsituents almost do not participate at all in the formation of the LUMO orbitals and remain electronically inactive. Therefore, the minimum of the LUMO energy profiles in series 2 and 4 consists of compounds with 2-substituents (i.e. 2.02, 2.04, 2.07 and 2.11) not having any neighboring interactions at the 3 position.
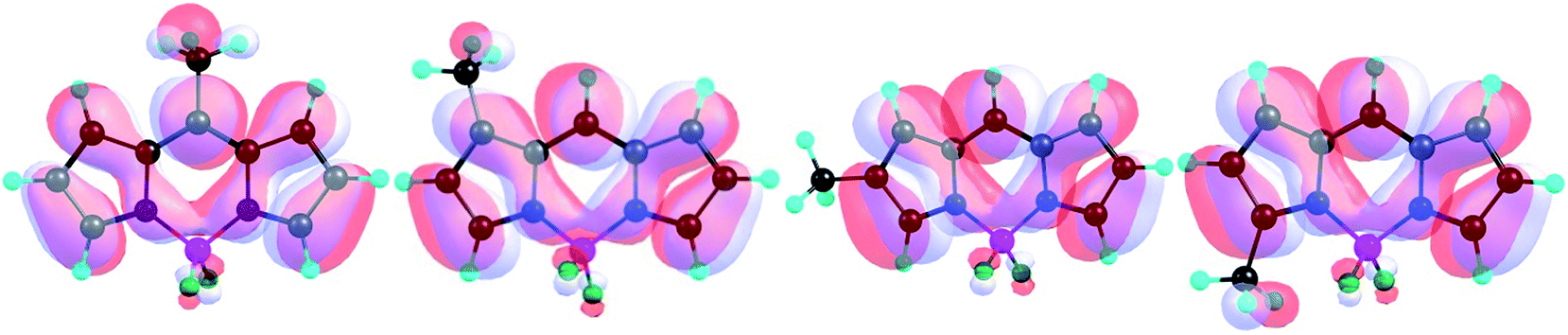 | ||
| Fig. 7 DFT B3LYP/6-31G(d)-obtained LUMOs of 1.01 to 1.04 (from left to right, respectively;, isovalue = 0.02). | ||
Effect on dihedral arrangement
The alkyl substitution patterns around the BODIPY units also actively participate in controlling the overall planarity of the fluorescent chromophore (see the ESI†). In order to understand such an effect, we considered the measurement of the dihedral angle between the two pyrrolic units (defined as ∠Py–Py), which can be taken as a standard of quantifying the planarity of BODIPY. If the dihedral angle is 0.0°, the system can be considered as planar, whereas larger values of this angle would indicate relatively more puckering of the BODIPY. As shown in Fig. 8 and 9, the methyl/ethyl substituents actively participate in controlling the planarity of the BODIPY core. | ||
| Fig. 8 Dihedral arrangements of the two neighboring pyrrolic units in compounds 1.01 to 1.04 (from left to right, respectively) as obtained from DFT B3LYP/6-31G(d) optimized ground-state structures. | ||
 | ||
| Fig. 9 Comparison of the dihedral arrangements of the two neighboring pyrrolic units in compounds in series 1 (top) and 2 (bottom) in their ground states (left) and in their 1st excited states (right), as obtained from DFT B3LYP/6-31G(d) computations. | ||
In series 1, from 1.01 to 1.03 the value of ∠Py–Py decreases gradually, and it takes a sudden drop on approaching 1.04. In fact, the BODIPY ring takes on a complete planar geometry (∠Py–Py = 0.000°) in 1.04, which is unexpected considering the possible steric effects. However, different observations are followed for series 3 and series 5, where 3.02 and 5.03 (respectively) show minimum puckering of the molecules (please see the ESI†). The tendencies observed in the 1st excited state optimized geometries of the BODIPY systems are in close similarity to the ground-state structures. As discussed, the 3 (and/or 5) alkyl substitutions in BODIPYs (Fig. 9 and ESI† for details) allow considerably minimal puckering of the ring structure in their ground states as well as upon electronic excitation. Such effects are intrinsically related to the total energies, i.e. relative stabilities, of the systems, and also contribute significantly to controlling conjugation through the molecular structures.
Collective comparison
The results presented earlier open many unexplored avenues related to the chemistry of BODIPY dyes. It is evident that even a small change in the random arrangement of an alkyl substituent can cause considerably large electronic and structural effects on the molecular systems. Although this study represents a qualitative outlook on the overall picture, it is evident that even such small changes cannot be made randomly if the synthetic designs are really expected to be built from the scratch. If alkyl substituents alone result in such large diversity, other electronically active atoms (e.g. Br, I, etc.) or functional entities would result in even larger effects on the molecular level, resulting in vastly different functionalities of closely related molecules.As observed, the alkyl substituents at the 3/5-positions of the BODIPY dyes participate in relative planarization of the molecular system, increasing conjugation throughout, which effectively results in its relatively higher stability compared to the other constitutional isomers. The results are unanimously supported from the observational consistencies of all four computational methods used in this work {i.e. B3LYP/6-31G(d), B3LYP/6-311++G(d,p), wb97xd/6-311++G(d,p) and mpw1pw91/6-311++G(d,p)}. The 3/5-positioned substituents also result in higher HOMO energies but give less effective destabilization of the LUMO levels, which results in an overall diminished band gap. These observations are line with the experimental observations as discussed vide-supra. The universality of the trends in isomeric molecules was tested using the four above mentioned computational methods (see also the ESI†) which unambiguously show the effect of alkyl groups in controlling the overall nature of BODIPYs.
Conclusions
In summary, a systematic approach has been utilized in order to achieve a comparative and qualitative perspective of BODIPYs with isomeric structures, differing only in substituent positions. The universality of the outcomes was also compared using different computational methods and the consistencies of any given trend were found to be unanimous in nature. The results found in this respect are potentially interesting and open new questions and prospects related to our available understanding of BODIPY-based molecules. Even alkyl moieties were found to be effective tools in altering the nature of BODIPY dyes and their intrinsic properties. It was found that the participation of such alkyl moieties in controlling the electronic signature of the BODIPYs (e.g. HOMO or LUMO energies) depends solely on the position of the substituent and is almost irrespective of the nature of substituent (e.g. methyl or ethyl groups). It was also found that the planarization or puckering of the BODIPY ring systems are also highly altered by the position of such alkyl substituents, which can be of great effect in the case of fluorescent chromophores. The results discussed in this work relate to the very basic and often puzzling experimental observations and can be of potential interest for the even finer tuning of the photophysics of BODIPY dyes.Acknowledgements
Sanjoy Mukherjee thanks CSIR, New Delhi for the Shyama Prasad Mukherjee Senior Research Fellowship. Thilagar thanks IISc and DST for financial support. The authors sincerely thank the IPC department, IISc for the computational facilities.References
- (a) A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891–4932 CrossRef CAS PubMed; (b) R. Ziessel, G. Ulrich and A. Harriman, New J. Chem., 2007, 31, 496–501 RSC; (c) G. Ulrich, R. Ziessel and A. Harriman, Angew. Chem., Int. Ed., 2008, 47, 1184–1201 CrossRef CAS PubMed; (d) A. C. Benniston and G. Copley, Phys. Chem. Chem. Phys., 2009, 11, 4124–4131 RSC; (e) N. Boens, V. Leen and W. Dehaen, Chem. Soc. Rev., 2012, 41, 1130–1172 RSC; (f) R. Ziessel and A. Harriman, Chem. Commun., 2011, 47, 611–631 RSC; (g) S. G. Awuaha and Y. You, RSC Adv., 2012, 2, 11169–11183 RSC; (h) H. Lu, J. Mack, Y. Yang and Z. Shen, Chem. Soc. Rev., 2014, 43, 4778–4823 RSC.
- (a) S. S. Agasti, A. M. Laughney, R. H. Kohler and R. Weissleder, Chem. Commun., 2013, 49, 11050–11052 RSC; (b) X. Zhang, C. Wang, L. Jin, Z. Han and Y. Xiao, ACS Appl. Mater. Interfaces, 2014, 6, 12372–12379 CrossRef CAS PubMed; (c) I. López-Duarte, T. T. Vu, M. A. Izquierdo, J. A. Bull and M. K. Kuimova, Chem. Commun., 2014, 50, 5282–5284 RSC; (d) C. Leong, S. C. Lee, J. Ock, X. Li, P. See, S. J. Park, F. Ginhoux, S.-W. Yun and Y.-T. Chang, Chem. Commun., 2014, 50, 1089–1091 RSC; (e) D. Collado, Y. Vida, F. Najera and E. Perez-Inestrosa, RSC Adv., 2014, 4, 2306–2309 RSC; (f) Z. Guo, S. Park, J. Yoon and I. Shin, Chem. Soc. Rev., 2014, 43, 16–29 RSC.
- (a) K. Krumova, L. E. Greene and G. Cosa, J. Am. Chem. Soc., 2013, 135, 17135–17143 CrossRef CAS PubMed; (b) A. Vázquez-Romero, N. Kielland, M. J. Arévalo, S. Preciado, R. J. Mellanby, Y. Feng, R. Lavilla and M. Vendrell, J. Am. Chem. Soc., 2013, 135, 16018–16021 CrossRef PubMed; (c) T. Wang, E. F. Douglass Jr, K. J. Fitzgerald and D. A. Spiegel, J. Am. Chem. Soc., 2013, 135, 12429–12433 CrossRef CAS PubMed; (d) R. Gotor, A. M. Costero, S. Gil, M. Parra, P. Gavina and K. Rurack, Chem. Commun., 2013, 49, 11056–11058 RSC; (e) S. Madhu, D. Kumar Sharma, S. K. Basu, S. Jadhav, A. Chowdhury and M. Ravikanth, Inorg. Chem., 2013, 52, 11136–11145 CrossRef CAS PubMed; (f) S. Madhu, R. Gonnade and M. Ravikanth, J. Org. Chem., 2013, 78, 5056–5060 CrossRef CAS PubMed; (g) E. Ganapathi, S. Madhu, T. Chatterjee, R. Gonnade and M. Ravikanth, Dyes Pigm., 2014, 102, 218–227 CrossRef CAS PubMed.
- (a) C. Y. Lee, O. K. Farha, B. J. Hong, A. A. Sarjeant, S. T. Nguyen and J. T. Hupp, J. Am. Chem. Soc., 2011, 133, 15858–15861 CrossRef CAS PubMed; (b) T. Bura, N. Leclerc, S. Fall, P. Lévêque, T. Heiser, P. Retailleau, S. Rihn, A. Mirloup and R. Ziessel, J. Am. Chem. Soc., 2012, 134, 17404–17407 CrossRef CAS PubMed; (c) R. Ziessel, G. Ulrich, A. Haefele and A. Harriman, J. Am. Chem. Soc., 2013, 135, 11330–11344 CrossRef CAS PubMed; (d) J. Min, T. Ameri, R. Gresser, M. Lorenz-Rothe, D. Baran, A. Troeger, V. Sgobba, K. Leo, M. Riede, D. M. Guldi and C. J. Brabec, ACS Appl. Mater. Interfaces, 2013, 5, 5609–5616 CrossRef CAS PubMed; (e) H. Yeo, K. Tanaka and Y. Chujo, Macromolecules, 2013, 46, 2599–2605 CrossRef CAS; (f) J.-F. Lefebvre, X.-Z. Sun, J. A. Calladine, M. W. George and E. A. Gibson, Chem. Commun., 2014, 50, 5258–5260 RSC.
- (a) L. Huang and J. Zhao, RSC Adv., 2013, 3, 23377–23388 RSC; (b) L. Huang, X. Cui, B. Therrien and J. Zhao, Chem. - Eur. J., 2013, 19, 17472–17482 CrossRef CAS PubMed; (c) C. Zhang, J. Zhao, S. Wu, Z. Wang, W. Wu, J. Ma, S. Guo and L. Huang, J. Am. Chem. Soc., 2013, 135, 10566–10578 CrossRef CAS PubMed; (d) S. Guo, H. Zhang, L. Huang, Z. Guo, G. Xiong and J. Zhao, Chem. Commun., 2013, 49, 8689–8691 RSC; (e) J. Ma, X. Yuan, B. Küçüköz, S. Li, C. Zhang, P. Majumdar, A. Karatay, X. Li, H. G. Yaglioglu, A. Elmali, J. Zhao and M. Hayvali, J. Mater. Chem. C, 2014, 2, 3900–3913 RSC; (f) P. Majumdar, X. Yuan, S. Li, B. Le Guennic, J. Ma, C. Zhang, D. Jacquemin and J. Zhao, J. Mater. Chem. B, 2014, 2, 2838–2854 RSC; (g) X. Cui, J. Zhao, Y. Zhou, J. Ma and Y. Zhao, J. Am. Chem. Soc., 2014, 136, 9256–9259 CrossRef CAS PubMed.
- A. D. Laurent, C. Adamo and D. Jacquemin, Phys. Chem. Chem. Phys., 2014, 16, 14334–14356 RSC.
- (a) O. Galangau, C. Dumas-Verdes, R. Meallet-Renault and G. Clavier, Org. Biomol. Chem., 2010, 8, 4546–4553 RSC; (b) H. Guo, Y. Jing, X. Yuan, S. Ji, J. Zhao, X. Lib and Y. Kanc, Org. Biomol. Chem., 2011, 9, 3844–3853 RSC; (c) G.-L. Fu, H. Pan, Y.-H. Zhao and C.-H. Zhao, Org. Biomol. Chem., 2011, 9, 8141–8146 RSC; (d) T. Sakida, S. Yamaguchi and H. Shinokubo, Angew. Chem., Int. Ed., 2011, 50, 2280–2283 CrossRef CAS PubMed; (e) H. Liu, J. Mack, Q. Guo, H. Lu, N. Kobayashi and Z. Shen, Chem. Commun., 2011, 47, 12092–12094 RSC; (f) N. Sakamoto, C. Ikeda and T. Nabeshima, Chem. Commun., 2010, 46, 6732–6734 RSC; (g) M. T. Whited, N. M. Patel, S. T. Roberts, K. Allen, P. I. Djurovich, S. E. Bradforth and M. E. Thompson, Chem. Commun., 2012, 48, 284–286 RSC.
- (a) S. Caprasecca, C. Curutchet and B. Mennuccia, Photochem. Photobiol. Sci., 2011, 10, 1602–1609 RSC; (b) J. Banuelos, F. L. Arbeloa, V. Martinez, M. Liras, A. Costela, I. G. Moreno and I. L. Arbeloa, Phys. Chem. Chem. Phys., 2011, 13, 3437–3445 RSC; (c) J.-L. Jin, H.-B. Li, Y. Geng, Y. Wu, Y.-A. Duan and Z.-M. Su, ChemPhysChem, 2012, 13, 3714–3722 CrossRef CAS PubMed; (d) B. L. Guennic, O. Maury and D. Jacquemin, Phys. Chem. Chem. Phys., 2012, 14, 157–164 RSC; (e) M. J. Calhorda, D. Suresh, P. T. Gomes, R. E. Di Paolo and A. L. Maçanita, Dalton Trans., 2012, 41, 13210–13217 RSC; (f) X. Liu, J. Zhang, K. Li, X. Sun, Z. Wu, A. Ren and J. Feng, Phys. Chem. Chem. Phys., 2013, 15, 4666–4676 RSC; (g) B. L. Guennic, S. Chibani, A. Charaf-Eddin, J. Massue, R. Ziessel, G. Ulrich and D. Jacquemin, Phys. Chem. Chem. Phys., 2013, 15, 7534–7540 RSC; (h) S. Chibani, A. Charaf-Eddin, B. L. Guennic and D. Jacquemin, J. Chem. Theory Comput., 2013, 9, 3127–3135 CrossRef CAS; (i) S. Chibani, A. Charaf-Eddin, B. Mennucci, B. L. Guennic and D. Jacquemin, J. Chem. Theory Comput., 2014, 10, 805–815 CrossRef CAS; (j) M. Buyuktemiz, S. Duman and Y. Dede, J. Phys. Chem. A, 2013, 117, 1665–1669 CrossRef CAS PubMed; (k) E. A. Briggs, N. A. Besley and D. Robinson, J. Phys. Chem. A, 2013, 117, 2644–2650 CrossRef CAS PubMed; (l) K. S. Radke, R. Scholz, F. Ortmann, K. Leo and G. Cuniberti, J. Phys. Chem. C, 2014, 118, 6537–6547 CrossRef CAS; (m) D. Jacquemin, S. Chibani, B. L. Guennic and B. Mennucci, J. Phys. Chem. A, 2014, 118, 5343–5348 CrossRef CAS PubMed; (n) P. Boulanger, D. Jacquemin, I. Duchemin and X. Blasé, J. Chem. Theory Comput., 2014, 10, 1212–1218 CrossRef CAS; (o) A. Charaf-Eddin, B. L. Guennic and D. Jacquemin, Theor. Chem. Acc., 2014, 133, 1456 CrossRef.
- (a) M. Baruah, W. Qin, N. Basarić, W. M. De Borggraeve and N. Boens, J. Org. Chem., 2005, 70, 4152–4157 CrossRef CAS PubMed; (b) E. Lager, J. Liu, A. Aguilar-Aguilar, B. Z. Tang and E. Pena-Cabrera, J. Org. Chem., 2009, 74, 2053–2058 CrossRef CAS PubMed.
- (a) C. A. Swamy, P. S. Mukherjee and P. Thilagar, Inorg. Chem., 2014, 53, 4813–4823 CrossRef PubMed; (b) S. Mukherjee and P. Thilagar, Chem. - Eur. J., 2014, 20, 9052–9062 CAS.
- C. A. Swamy, P. S. Mukherjee and P. Thilagar, Chem. Commun., 2013, 49, 993–995 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- (a) A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS; (b) C. Lee, W. Yang and R. G. Parr, s. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS; (c) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS PubMed.
- J. D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- C. Adamo and V. Barone, J. Chem. Phys., 1998, 108, 664–675 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra12071j |
| This journal is © The Royal Society of Chemistry 2015 |