
A non-immunosuppressive FK506 analogue with neuroregenerative activity produced from a genetically engineered Streptomyces strain†
Abstract
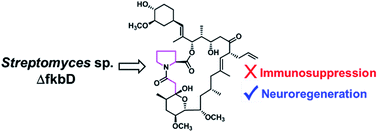
FK506 exhibits neuroprotective and neuroregenerative activities in addition to its clinically important immunosuppressant properties. The macrolide ring of FK506 is biosynthesized by a hybrid polyketide synthase/nonribosomal peptide synthetase system and is further modified via C-9 oxidation by FkbD and 31-O-methylation by FkbM. A new FK506 analogue, 9-deoxo-prolyl-FK506 (1), was isolated from the fkbD deletion mutant of Streptomyces sp. KCTC11604BP, and its biological activities were evaluated. The in vitro immunosuppressive activity was significantly reduced, but in vitro neurite outgrowth activity similar to FK506 was maintained. These results demonstrate the potential of pathway engineering for the modification of structurally complex natural products, such as FK506, to create improved biological agents.
 Please wait while we load your content...
Please wait while we load your content...

