
An α1-adrenergic receptor ligand repurposed as a potent antiproliferative agent for head and neck squamous cell carcinoma†

Abstract
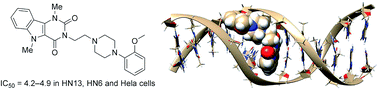
In this study we report the anticancer properties of RN5-Me, an α1-adrenergic receptor ligand. Biological screening and circular dichroism data indicate that it acts as a DNA intercalator. Docking studies, confirming this behaviour, suggest that RN5-Me possesses great selectivity for alternating AT nucleobases upon GC ones. In the cytotoxicity assay, it shows IC50 values in the range of 4.2–25.0 nM towards the cancer cell lines HN6, HN13, HeLa, SK-Me1-103, PC3, and MCF7. It is noteworthy that RN5-Me shows a higher selectivity than Cisplatin for HN6 and HN13 over NOK-SI non-cancerous cells.
 Please wait while we load your content...
Please wait while we load your content...

