
Tuning the work function of polyaniline via camphorsulfonic acid: an X-ray photoelectron spectroscopy investigation
Omar Abdulrazzaq*a,
Shawn E. Bourdo*a,
Viney Sainia,
Fumiya Watanabea,
Bailey Barnesa,
Anindya Ghoshb and
Alexandru S. Birisa
aCenter for Integrative Nanotechnology Sciences, University of Arkansas at Little Rock, Little Rock, AR, USA. E-mail: oaabdulrazza@ualr.edu; sxbourdo@ualr.edu; Fax: +1-501-683-7601; Tel: +1-501-683-7456
bDepartment of Chemistry, University of Arkansas at Little Rock, Little Rock, AR, USA
First published on 19th November 2014
Abstract
In this work, we present the first demonstration of tuning the work function of polyaniline by controlling the concentration level of camphorsulfonic acid as a protonic acid dopant and m-cresol as a solvent. Optical, thermal, structural, and electronic properties, along with surface topography and elemental analysis of protonated polyaniline, were studied in detail to investigate the effect of camphorsulfonic acid on the work function of polyaniline. The results showed that an increase in camphorsulfonic acid content induces a gradual transformation in the polyaniline structure from an emeraldine base to an emeraldine salt phase, which is associated with an increase in electrical conductivity and an improvement in crystallinity. X-ray photoelectron spectroscopy was used to evaluate the work function and to determine the elemental composition of the surface and several atomic layers beneath the surface. The results showed that increasing the camphorsulfonic acid content from quarter protonated to fully protonated leads to an increase in the work function of polyaniline from 4.42 ± 0.14 eV to 4.78 ± 0.13 eV.
Introduction
Recently, a significant effort has been directed toward the development of conjugated polymer systems in order to establish the basis of third-generation of photovoltaics, i.e., organic solar cells (OSCs).1–3 OSCs are economical, flexible, lightweight, and easy to fabricate especially on a large scale.4 However, there are critical barriers to the commercial production of OSCs: degrading performance as a function of time, environmental stability, and low efficiency compared to the first and second generations of solar cells.1 A considerable amount of research has been performed to overcome the difficulties associated with OSCs, e.g., finding an alternative to the most widely used hole transport layer (HTL): poly(3,4-ethylenedioxythiophene):polystyrene sulfonate (PEDOT:PSS).5 PEDOT:PSS thin film is used in OSCs to boost the hole injection into the anode, as well as to smooth the ITO surface in the standard (or non-inverted) OSCs. Despite the fact that PEDOT:PSS solar cells have been fine-tuned, several problems with PEDOT:PSS have been reported to be a critical barrier. PEDOT:PSS is a hygroscopic material and can degrade easily when exposed to water or water vapor, and it can also act as a reservoir for water that in turn allows water to diffuse in the inner layers of the device and react with other components.6 Researchers have found that PEDOT:PSS solar cells degrade rapidly even when the cells are encapsulated.7 This is primarily due to the hygroscopic nature and acidity of PEDOT:PSS film, but also is a result of the migration of PSS to the adjacent layers, which can lead to the deterioration of solar cell performance.6 Several alternatives for PEDOT:PSS have been suggested in the literature: polyaniline,5 nickel oxide,8 and graphene oxide.9 In order for OSCs to perform efficiently, they require proper energy band alignment between ITO, HTL, and HOMO (highest occupied molecular orbital) of the polymer (electron donor) to accomplish a low voltage drop for injection of holes.10,11 ITO work function is reported to have values from 3.8 eV to 5.1 eV depending on the surface cleaning and treatment method.12 In addition, the HOMO level varies based on the electron donating polymer composition.13,14 Considering all of these variables, it is essential to find a HTL with a work function that can be tuned easily to achieve a proper alignment with the adjacent layers.Recent studies have focused on a more complicated architecture called tandem cells, a stack of two or more subcells, in order to boost the performance of OSCs. The tandem architecture is designed to absorb over a wider spectral range, since it has two or more subcells—each of which is made of a different polymer that has an optical band gap in a different region of the solar spectrum. This system is reported to achieve a power conversion efficiency of up to 10%.15 One of the major challenges in the fabrication of tandem organic solar cells is the design of the recombination interlayer (the buffer layer that interconnects the front subcell with the back subcell). Similar to the energy level alignment of HTL (or ETL) in single junction OSCs, in order to obtain optimum power conversion efficiency, the recombination layer must have a properly aligned work function so that electrons from the front cell and holes from the back cell can flow into the layer then recombine. Commonly, two different materials are used as recombination interlayers, usually by modifying one interface to be the high work function side and the other to be the low work function side.16 PEDOT:PSS has been used frequently as a recombination interlayer by modifying one side to be the low work function value side for electron collection.16,17 One of the disadvantages of using PEDOT:PSS in tandem geometry is that it is applied from an aqueous solution that can lead to degradation of the front subcell. Other researchers have used evaporation methods to deposit metals such as gold or silver.18 However, this technique can be hampered by the high deposition temperature that can degrade the front subcell and is not economical due to its high fabrication costs. Solution-processed interlayers that dissolve the layer beneath (i.e., the front subcell) must be avoided.17 Therefore, interlayers should be made in such a way that—in addition to matching the work function with the upper and bottom subcells—they are processed from a solvent that is not aqueous and does not cause the active layer underneath to dissolve.
Polyaniline (PANI) is one of the most frequently investigated conducting polymers for many reasons: it is environmentally stable, inexpensive, and its electrical conductivity can be easily tuned.19 PANI is a conjugated polymer that can serve as an insulator, semiconductor, or conductor, depending on the degree of protonation.11 Generally, emeraldine base is the insulating phase of PANI with two amine groups (–N–) and two imine groups (![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–), per repeat unit. The doping (protonation) of PANI with a protonic acid converts some or all of the imine groups into amine groups via internal redox reaction resulting in another phase of PANI, emeraldine salt, which is conductive. PANI can be dissolved using solvents such as chloroform, xylene, and/or m-cresol.20–22 Each solvent can have a certain degree of effect on the film morphology and surface topography of PANI thin films after processing from such solvents.20 For example, PANI cast out of chloroform exhibits localized polarons in short conjugated lengths owing to the compact coil conformation. In contrast, PANI cast from m-cresol exhibits a transition between energy bands within the half-filled delocalized polaron band that can be observed in the UV-Vis-NIR measurements as a free carrier tail, a feature of planar or expanded coil conformation.20 MacDiarmid and Epstein have defined this phenomenon as a “secondary doping”.22 Various dopants have been used to protonate PANI, such as hydrochloric acid (HCl) or camphorsulfonic acid (CSA).19,23 It has been demonstrated that the dopant type24 and dopant concentration11 can play a significant role in determining the structural properties of PANI films. In our previous study, we found that increasing the CSA protonation level results in an improvement in PANI structure by increasing the interchain stacking of PANI chains,11 while further protonation (such as overprotonation) could result in a decrease in the crystallinity of PANI because excessive CSA may induce a crosslinking effect. It has been postulated by MacDiarmid and Epstein that CSA together with m-cresol has a great effect on the chain stacking because m-cresol promotes more dissolution or solvation of the positive and negative charges on the polymer chain and the negative charges of CSA anions. This will inhibit the interaction between positive and negative charges at the same chain prompting more expanded coil conformation.22
N–), per repeat unit. The doping (protonation) of PANI with a protonic acid converts some or all of the imine groups into amine groups via internal redox reaction resulting in another phase of PANI, emeraldine salt, which is conductive. PANI can be dissolved using solvents such as chloroform, xylene, and/or m-cresol.20–22 Each solvent can have a certain degree of effect on the film morphology and surface topography of PANI thin films after processing from such solvents.20 For example, PANI cast out of chloroform exhibits localized polarons in short conjugated lengths owing to the compact coil conformation. In contrast, PANI cast from m-cresol exhibits a transition between energy bands within the half-filled delocalized polaron band that can be observed in the UV-Vis-NIR measurements as a free carrier tail, a feature of planar or expanded coil conformation.20 MacDiarmid and Epstein have defined this phenomenon as a “secondary doping”.22 Various dopants have been used to protonate PANI, such as hydrochloric acid (HCl) or camphorsulfonic acid (CSA).19,23 It has been demonstrated that the dopant type24 and dopant concentration11 can play a significant role in determining the structural properties of PANI films. In our previous study, we found that increasing the CSA protonation level results in an improvement in PANI structure by increasing the interchain stacking of PANI chains,11 while further protonation (such as overprotonation) could result in a decrease in the crystallinity of PANI because excessive CSA may induce a crosslinking effect. It has been postulated by MacDiarmid and Epstein that CSA together with m-cresol has a great effect on the chain stacking because m-cresol promotes more dissolution or solvation of the positive and negative charges on the polymer chain and the negative charges of CSA anions. This will inhibit the interaction between positive and negative charges at the same chain prompting more expanded coil conformation.22
Work function (WF), the minimum work required to remove an electron from the surface of a solid, is an inherent property of solid surfaces. The work function for a certain material can differ depending on the electronic structure, and also on the surface orientation.25 Furthermore, work function is affected by surface contamination and roughness.26,27 WF can be determined using techniques such as ultraviolet photoemission spectroscopy (UPS) and X-ray photoelectron spectroscopy (XPS).26 In the XPS technique, the extreme ends of the spectra are acquired so that cutoffs at both low and high binding energies can be displayed. To avoid the interference between instrument and sample work functions, the sample should be biased before collecting data. Next, the whole spectrum must be shifted so that the Fermi level is aligned with zero eV binding energy. Unlike some other common techniques, such as the Kelvin probe method that only gives a relative value of work function, XPS and UPS techniques can provide an absolute work function value.
Thus far, very few studies have investigated the work function of PANI.21,28–31 The majority of these studies have made use of relative methods by measuring the difference between the potential of the sample and a reference electrode. Relative methods can provide the relative WF variation without identifying an absolute value, which is essential when designing organic solar cells. Jinsung et al. found that, by increasing the HCl concentration in PANI nanoparticles, the WF decreases by a value up to 0.27 eV.30 Polk et al. investigated the effect of some strong protonic acids, such as triflic acid, on the WF of PANI, and showed that the relative WF decreases with increasing acid concentration. The phenomenon was attributed to the formation of localized states after protonation that could cause the WF to decrease.29 Ahmed et al. investigated temperature-sensing polyaniline films doped with three ratios of CSA and used m-cresol as the solvent, with no significant effect on the WF of PANI.31 Chinn et al. studied the change in the relative WF of doped and undoped PANI when exposed to moisture. They observed that the WF of PANI increased rapidly after exposure to 100% relative humidity using nitrogen as a carrier gas. They also reported that the WF rapidly recovered after the humidity was removed.28 Posdorfer et al. demonstrated that the WF of PANI:PSS cast from a water-based solution increases from 4.75 to 5.06 eV after increasing the PSS dopant (polystyrene sulfonate), and with heavy doping the WF of PANI lowers from 5.06 to 4.88 eV.21 By studying the WF using different solvents, they found a linear relationship between the WF of PANI and the dielectric constant of the solvent. However, based upon previous studies in which different researchers present varying results, it is not clear how doping affects the WF of polyaniline. Therefore, it is worth performing a comprehensive study to assess the absolute value of the work function of polyaniline and its relationship with various protonation levels using CSA as a dopant and m-cresol as a solvent. Such a study is very important in order to find criteria for tuning the WF of non-aqueous PANI for further use as a HTL and/or a recombination layer in organic tandem solar cells. The choice of CSA and m-cresol with PANI is important with respect to specific applications such as HTL in organic solar cells or organic light-emitting diodes because, in such applications, it is preferable to cast HTL from non-aqueous solutions. It is also preferable for PANI to be in the expanded coil conformation rather than in the compact coil conformation. The expanded conformation introduces higher conductivity and can provide better pathways for holes to transport through the HTL to the anode.
Our results on PANI:CSA as a HTL in organic solar cells have been previously published.11 In the present article, we report an approach for determining the WF of polyaniline using XPS technique. The main thrust of this work is to tune the work function of polyaniline via protonation with various doping levels of camphorsulfonic acid. The findings have potential for further use in organic tandem solar cells in which the work function of hole transport, electron transport, and recombination layers is crucial. The bedrock of utilizing XPS for work function measurements of metals and alloys has been previously established.32 Accordingly, we embraced this technique to determine the work function of polyaniline. This detailed study includes optical, electrical, structural, elemental, and electronic properties of PANI:CSA. The investigation is designed to obtain a better understanding of the material and the reasons behind the change in the WF of PANI with various CSA doping contents.
Experimental procedure
Polyaniline was synthesized by using oxidative polymerization of aniline in acidic solution as described by MacDiarmid.19 Details of the synthesis can be found elsewhere.11 In brief, 2 mL of double-distilled aniline were added to 50 mL of 1 M methane sulfonic acid and cooled to ∼0 °C. Next, 5.24 g of sodium persulfate were added to the solution and kept in constant stirring for 6 hours. The resulting precipitate (dark green) of doped polyaniline was vacuum-filtered and washed with deionized water then partially de-doped by washing with 15 mL of 1 M ammonium hydroxide. A thorough de-doping was carried out by overnight stirring in a 0.1 M aqueous ammonium hydroxide. The final step was vacuum-filtering the product and washing with acetone to remove any oligomers and intermediate impurities.Following the synthesis, four different solutions were prepared with varied molar ratios of CSA. Quarter-protonated PANI solution (denoted as QP-PANI) was prepared by mixing 72.4 mg of PANI with 23.2 mg of CSA very well using mortar and pestle, adding to a vial, and finally adding 4.78 g of m-cresol. (Improper mixing of PANI with CSA in the mortar and pestle could result in a jelled solution.) The mixture was then homogenized immediately at 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 30 min using a high speed mixer (T 18b ULTRA-TURRAX S1 with S 18N-10G attachment). Any delay in transferring the mixture to the homogenizer could cause the powder to accumulate as undissolved chunks in the solution. After homogenizing, the solution was magnetically stirred for at least 24 hours then centrifuged at 3500 rpm for 30 min to remove any insoluble components. In the final stage, the supernatant was decanted and kept under constant stirring until use. Similarly, half-protonated PANI (HP-PANI), three quarters-protonated PANI (3QP-PANI), and fully-protonated PANI (FP-PANI) were prepared by dissolving 72.4 mg of PANI and 46.5 mg of CSA in 5.95 g of m-cresol, 72.4 mg of PANI and 69.2 mg of CSA in 7.1 g of m-cresol, and 72.4 mg of PANI and 93 mg of CSA in 8.27 g of m-cresol, respectively. The above PANI and CSA masses were chosen to attain the required molar ratios, i.e., QP-PANI consists of one CSA molecule alongside two PANI units; HP-PANI is one CSA molecule alongside one PANI unit; 3QP-PANI consists of three CSA molecules alongside two PANI units; and FP-PANI consists of two CSA molecules alongside one PANI unit. The mass of the solvent was calculated to produce a solution of 2% w/w of PANI:CSA in m-cresol.
000 rpm for 30 min using a high speed mixer (T 18b ULTRA-TURRAX S1 with S 18N-10G attachment). Any delay in transferring the mixture to the homogenizer could cause the powder to accumulate as undissolved chunks in the solution. After homogenizing, the solution was magnetically stirred for at least 24 hours then centrifuged at 3500 rpm for 30 min to remove any insoluble components. In the final stage, the supernatant was decanted and kept under constant stirring until use. Similarly, half-protonated PANI (HP-PANI), three quarters-protonated PANI (3QP-PANI), and fully-protonated PANI (FP-PANI) were prepared by dissolving 72.4 mg of PANI and 46.5 mg of CSA in 5.95 g of m-cresol, 72.4 mg of PANI and 69.2 mg of CSA in 7.1 g of m-cresol, and 72.4 mg of PANI and 93 mg of CSA in 8.27 g of m-cresol, respectively. The above PANI and CSA masses were chosen to attain the required molar ratios, i.e., QP-PANI consists of one CSA molecule alongside two PANI units; HP-PANI is one CSA molecule alongside one PANI unit; 3QP-PANI consists of three CSA molecules alongside two PANI units; and FP-PANI consists of two CSA molecules alongside one PANI unit. The mass of the solvent was calculated to produce a solution of 2% w/w of PANI:CSA in m-cresol.
PANI thin films with different doping concentrations were prepared by spin coating the four solutions on ITO glass (NaOH–water–IPA treated/cleaned) at 2000 rpm, followed by drying in the ambient atmosphere at 60–70 °C for 1–2 hours before storing them in a vacuum desiccator. ITO cleaning is a vital step in obtaining uniform PANI films. The detailed procedure for treating and cleaning ITO substrates has been described in our previous report.11
A Fourier transform infrared (FTIR) spectrometer (Thermo Scientific Nicolet 6700 equipped with Smart iTR™ Attenuated Total Reflectance Accessory) was used to record the infrared spectra of the product. Thickness of the films was determined by using a Horiba Jobin Yvon UVISEL ellipsometer by fitting Ψ (amplitude ratio) and Δ (change in phase) ellipsometric data with the aid of DeltaPsi2 software environment. The thickness of all of the samples was determined to be between 30–50 nm. A 100 nm thick layer of silver was vacuum-evaporated on part of the surface for each film using a shadow mask. Absorption spectra of the films in the range of 300–900 nm were collected by a UV-Vis-NIR double beam spectrophotometer (Shimadzu UV-3600). The Hall effect measurements were performed on PANI:CSA films using an Ecopia HMS-5000 Hall effect system at room temperature. In Hall measurements, thicker films of PANI:CSA (ca. 100 nm) were cut into 1 cm × 1 cm, and silver paste was applied to the corners in a Van der Pauw configuration. The Hall terminals were placed at the silver contact, and Hall voltages were directly measured at a magnetic flux density of 0.55 T and six different current flows ranging from 0.1 mA to 1.0 mA. The thermal properties of freestanding PANI:CSA films were obtained by thermogravimetric analysis (TGA) using METTLER TOLEDO TGA/SDTA851. TGA was carried out from 25 to 850 °C at the rate of 5 °C min−1 under air. X-ray diffraction (XRD) was performed on the samples using Bruker D8 Discovery. Surface topography and roughness were measured in tapping mode using a Veeco Dimensions 3100 Atomic Force Microscope (AFM) at a 10 μm × 10 μm scale. The AFM tip was brought down on a 1 μm × 1 μm scan at 1 Hz and 256 lines to focus the integral gain, proportional gain, and amplitude setpoint parameters. Once a good image was achieved, the scan was set to 10 μm × 10 μm at 0.1 Hz and 512 lines. Every image was flattened initially. Roughness analysis and surface area were performed for each scan. Each film was scanned in three different locations, and the roughness values were averaged to represent the surface morphological properties of the film. Elemental analyses were carried out by performing a survey scan using X-ray photoelectron spectroscopy type Thermo Scientific K-Alpha instrument with monochromated Al Kα excitation source with photon energy 1486.6 eV (36 W maximum power). The X-ray spot size was set to 400 μm and pass energy of 200 eV. The same instrument was used to estimate the work function of PANI:CSA films by using a work function stage which is capable of biasing the sample (Thermo Scientific K-Alpha Work Function Module). The stage also includes a mounted gold foil standard for WF value calibration purposes. In the after-etching measurements of work function, the ion beam energy was set to 1000 eV, the etching time to 10 s, and the current was set at low. The spectra at both high and low binding energy ends were acquired so that cutoffs at both ends of the binding energies could be introduced. Samples were biased at −30 V in order to avoid the effect of the instrument work function. The sample spectra were then shifted so that the Fermi level (onset of electron emission) was aligned with zero eV binding energy. The difference between the photon energy of Al Kα (1486.6 eV) and the high binding energy cutoff (i.e., the cutoff of the kinetic energy of the photoelectron) yields the work function of the sample. Fig. 1 is an example of data obtained from a gold foil standard; the WF was found to be 5.1 ± 0.1 eV. The XPS measurements mentioned above were averaged from a total of 20 scans for each single measurement to ensure the accuracy of the results.
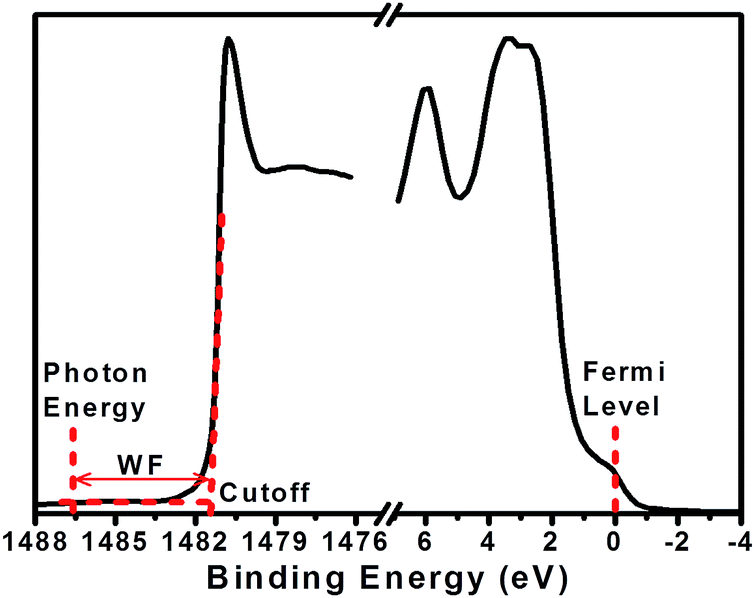 | ||
| Fig. 1 The two parts of the survey spectrum, i.e., low and high binding energy regions (Fermi and cutoff scan) acquired from XPS with a monochromatic radiation from Al Kα for gold foil standard reference as an example to calculate the work function. | ||
Results and discussion
Fig. 2 displays the FTIR spectrum of the synthesized PANI powder to confirm the chemical structure. All peaks of PANI recognized from the transmittance spectrum were typical of emeraldine base polyaniline.33–35 The bands with peak maximum at wavenumbers 825, 1159, 1214, 1300, 1499, 1590, and 3382 cm−1 were assigned to out-of-plane C–H bending, in-plane C–H bending, C–N bending, amine C–N stretching, CC stretching of benzenoid, CC stretching of quinoid, and N–H stretching, respectively. The weak peak of O–H stretching at 3270 cm−1 was assigned to water which is due to adsorption of moisture after polymerization.
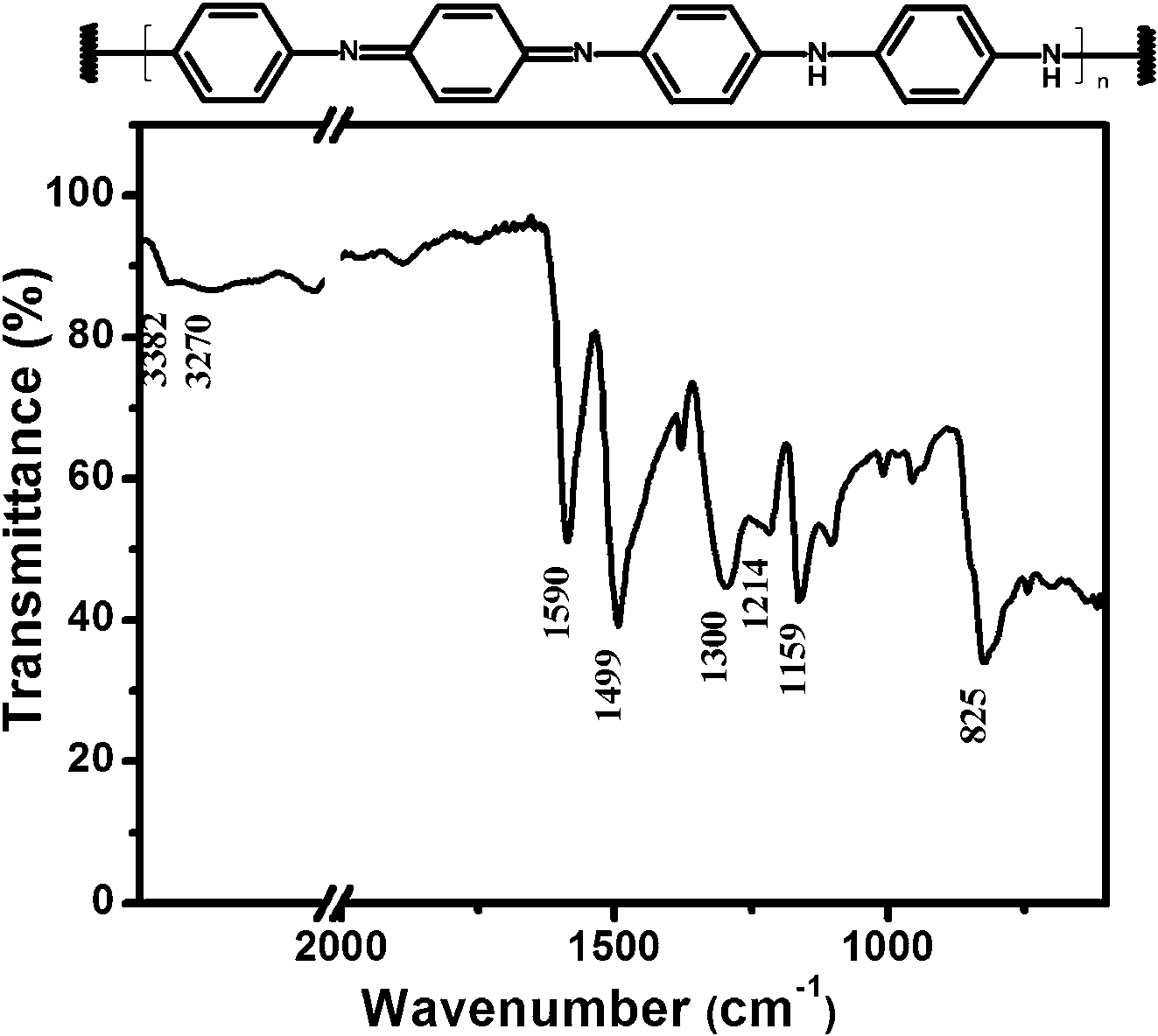 | ||
| Fig. 2 Chemical schematic and FTIR spectrum of emeraldine base PANI synthesized by MacDiarmid method.19 | ||
In order to precisely determine the protonation levels of the films, elemental analysis was performed using XPS. The survey scans of PANI:CSA thin films are plotted in Fig. 3(a). The plots show peaks at binding energies 164.3 eV, 285.2 eV, 399.8 eV, and 532.4 eV, which correspond to S2p, C1s, N1s, and O1s, respectively.36 Fig. 3(b) shows the enlarged peaks of sulfur and nitrogen, and the sulfur to nitrogen (S/N) atomic ratios for an average of three different measurements are shown in Fig. 3(c). Comparing the desired protonation levels (i.e., QP-, HP-, 3QP-, and FP-PANI) that were massed out in the experimental section with the XPS data, we can see that the values of the molar ratio (based on weight calculations) are not perfect but are close to the atomic ratio (determined from XPS measurements). Based on the molar ratio (from weighing calculations), the S/N of QP-PANI is 0.25, HP-PANI is 0.50, 3QP-PANI is 0.75, and FP-PANI is 1.00, while the average of the atomic ratios (from XPS measurements) is 0.31 for QP-PANI, 0.48 for HP-PANI, 0.62 for 3QP-PANI, and 0.84 for FP-PANI. The discrepancy between molar and atomic ratios is partially due to any incomplete dissolution of the PANI or CSA in m-cresol and/or due to weight loss of materials that stick to the vial wall, weighing paper, homogenizer rod, centrifuging, and mortar & pestle during grinding. Although the S/N ratios extracted from XPS are more realistic, we will continue to use the molar nomenclature while describing the experiment throughout this article since the atomic ratio can vary slightly during sample preparation.
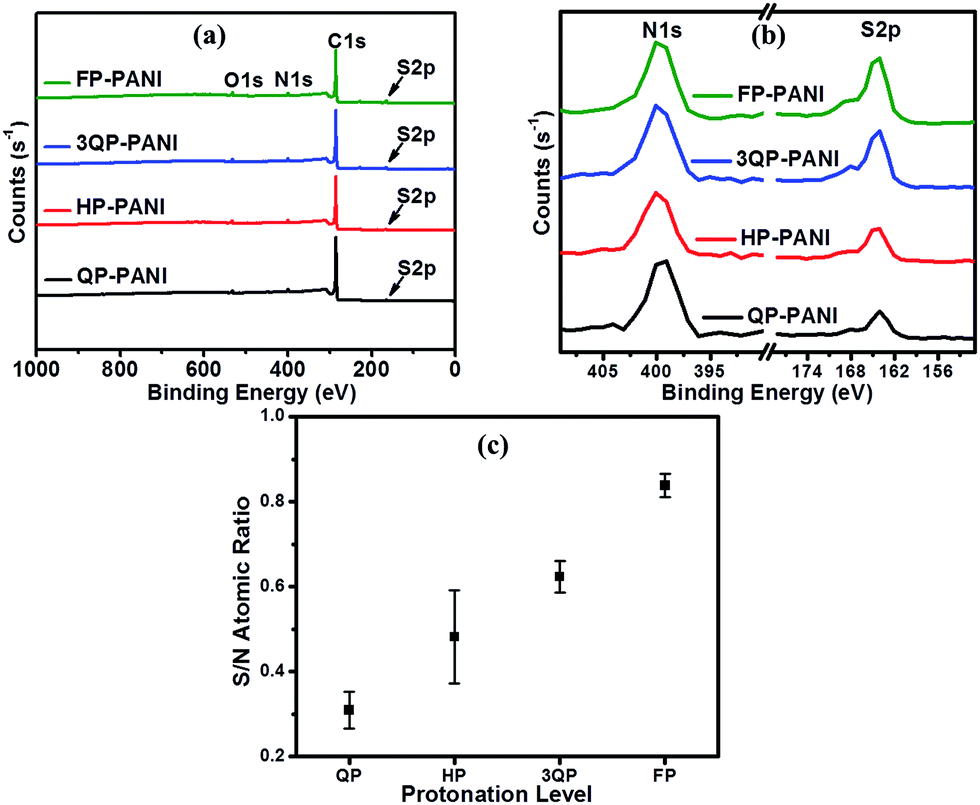 | ||
| Fig. 3 (a) Survey scan for PANI protonated with four different CSA ratios, (b) enlarged peaks of sulfur and nitrogen of PANI:CSA films, and (c) variation of sulfur to nitrogen atomic ratio of PANI:CSA films. | ||
The TGA plots in Fig. 4(a) show that the thermal stability of PANI has decreased with increasing CSA content probably due to the differences in morphology and reactivity of the polymer chain caused by the doping process. Similar results for PANI:CSA on thermal stability have been reported in the literature.37 All four polymers were observed to have a three-stage mass loss as is illustrated in the derivative curves shown in the inset of Fig. 4(a). The first mass loss is observed over a temperature range ca. 100–200 °C and is attributed to the evaporation of moisture, residual solvent, and the degradation of low molecular weight oligomers.38 However, less weight loss is observed when the protonation level increases. The second weight loss is associated with the thermal de-doping process of PANI39 while the third weight loss in the range of ca. 508–525 °C is due to the degradation of polymer chains.38 The midpoint of the combustion temperature of PANI:CSA films is presented in Fig. 4(b) for a direct comparison. As we can see, the PANI:CSA main chain degradation temperature noticeably decreases with increasing CSA protonation levels, which can be attributed to a more reactive polymer.
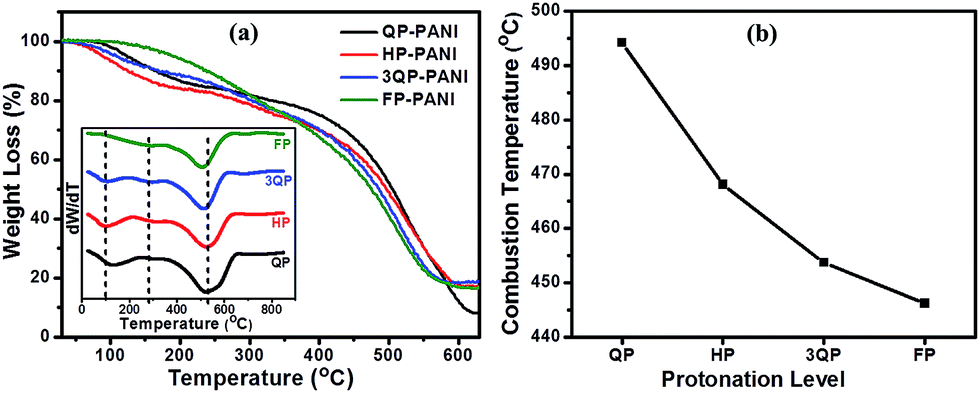 | ||
| Fig. 4 (a) TGA curves of PANI films doped at various CSA protonation levels, and (b) midpoint of the combustion temperature of PANI films doped at various CSA protonation levels. The inset shows the derivatives of the curves in graph (a). | ||
The optical absorption spectra of PANI:CSA thin films are illustrated in Fig. 5(a) and show two distinctive peaks. The peak at ca. 340 nm is related to the π–π* transition of benzenoid rings which becomes much weaker with increasing CSA protonation levels due to the formation of new energy bands. The peak at ca. 450 nm is assigned for polaron–π* and becomes stronger with increasing CSA protonation. The steady increase in the near-IR region of 3QP-PANI and FP-PANI films is associated with the free-carrier tail (delocalized polaron) that arises due to transformation from compact coil to expanded coil conformation.40 These dramatic changes in absorption spectra confirm that CSA has a significant influence on the conformation of the chains, which improves the crystallinity and increases the electrical conductivity by enhancing the interaction between the adjacent chains and reducing the twist defects between the aromatic rings.
 | ||
| Fig. 5 (a) UV-Vis absorption spectra of different PANI:CSA protonation levels, and (b) XRD patterns of different PANI:CSA protonation levels. | ||
The XRD patterns of PANI films protonated with various levels of CSA are presented in Fig. 5(b). These diffraction patterns exhibit clear differences associated with increased protonation levels. In order to illustrate the evolution of crystallinity from an undoped emeraldine base PANI (zero-protonation, ZP-PANI) to FP-PANI, the XRD patterns are shown in Fig. 5(b). Both ZP- and QP-PANI samples exhibit an amorphous pattern with a broad peak at 2θ ∼17.5°, and this is attributed to a compact coil conformation.41 Therefore, the QP-PANI has poor crystallinity that can be said to be structurally similar to that of the emeraldine base polyaniline,41 obviously owing to the low level of protonation. The peak of this sample corresponds to the (110) plane of the emeraldine base PANI. In the higher protonated films (i.e., HP-PANI, 3QP-PANI, and FP-PANI samples), the observed peaks at ∼15.5°, 20°, and 25.5° correspond to the (010), (100), and (110) planes of emeraldine salt PANI, respectively.42 The peak at ∼17.5° (observed in QP-PANI) becomes nearly indistinguishable in the higher protonated samples. Instead, three peaks become more dominant in the higher protonated films, implying better face-to-face interchain stacking between phenyl rings43 and therefore a more expanded coil conformation. These peaks are a result of the induced crystallinity of PANI by CSA and show that PANI chains in the PANI:CSA films have a more ordered structure since CSA enhances short-range order by increasing the interchain stacking of PANI chains.41,44 In addition, the origin of a small peak at 2θ ∼6.5° in higher protonated samples is claimed by Khalid et al. to be assigned to the periodic distance between the dopant and the N atom on adjacent main chains.45
To study the effect of PANI protonation level on the surface topography, AFM was utilized to determine the rms surface roughness. As seen in Fig. 6, a higher roughness is observed at lower protonation levels. The observed decrease in roughness with increased protonation is likely due to the transition from compact coil to expanded coil conformation. However, HP-PANI exhibits greater roughness than QP-PANI. Therefore, the surface area of these samples was also investigated to complement the rms roughness findings in order to more fully describe the surfaces. The mean value of the surface area determined by AFM (on 3 separate spots) was as follows: 101.79 ± 1.17 μm2, 101.17 ± 0.17 μm2, 100.19 ± 0.10 μm2, and 100.11 ± 0.01 μm2 for QP-PANI, HP-PANI, 3QP-PANI, and FP-PANI, respectively. Unlike rms roughness, the surface area of QP-PANI is higher than that of HP-PANI. This suggests that even though rms roughness of HP-PANI is higher than that of QP-PANI, the number of spikes per unit area in the QP-PANI image is greater than that of HP-PANI. This observation of a much lower surface roughness with increasing protonation level is a result of an expanded coil conformation where the polymer chains lie flat instead of tangling. Any future work on using PANI:CSA as a recombination layer in organic tandem solar cells may require a smoothing of the surface for the lower protonation levels of doped PANI to improve the performance of the cells. Lodha et al.38 found that PANI films made out of N,N′-diemthyl propylene urea solvent and doped by immersing the films in HCl showed an increase in rms roughness up to 46% after doping. This may indicate that PANI roughness is strongly dependent on solvent type and doping method.
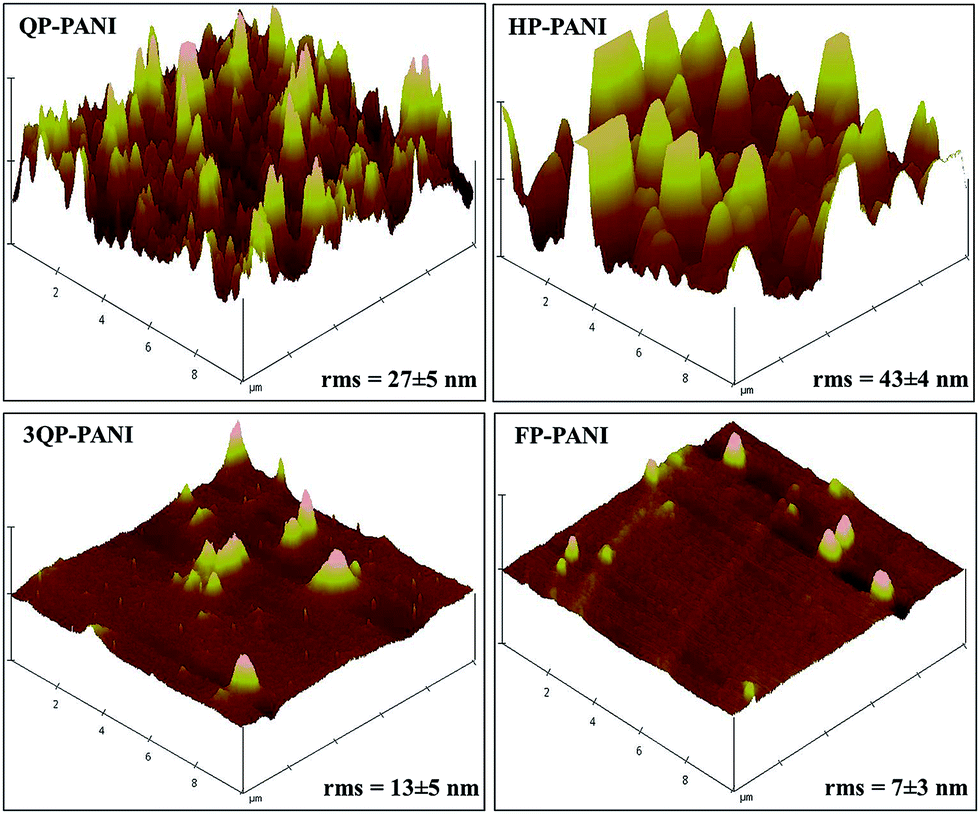 | ||
| Fig. 6 AFM images of PANI protonated with various CSA levels. | ||
The electrical conductivity of various PANI:CSA films—which increases up to two orders of magnitude when CSA protonation is increased from quarter to fully protonated—is displayed in Fig. 7(b). The further increase in conductivity is attributed to the improvement in crystallinity [Fig. 5(b)] and the phase change from compact to expanded coil conformation which in turn increases the delocalized carriers. It is well-known that there is a strong correlation between crystallinity and conductivity of a conducting polymer.46 An expanded coil conformation allows more interaction between adjacent chains and also results in the polaron band having a wider dispersion in energy leading to a higher electrical conductivity.40 A previous study suggested that PANI:CSA with a protonation level of 30% lies at the metal-insulator boundary conduction mechanism.47 In our case, QP-PANI represents a protonation level of 25% molar ratio (31% S/N atomic ratio) which is close to that boundary, and this assumption is supported by the behavior illustrated in Fig. 6(a), where a dramatic decrease in the intensity of π–π* transition peak for HP-, 3QP-, and FP-PANI films is evident. This decrease is associated with an increase in the polaron–π* transition peak because the band gap between the π band and the polaron band is eliminated, and, consequently, polarons become more delocalized.40 These changes in electronic structure can explain the noticeable increase in conductivity (2 S cm−1 for QP-PANI to more than 200 S cm−1 for FP-PANI). To support the assumption about the increase in the delocalized carriers, the concentration of free carriers as a function of protonation level is presented in Fig. 7(b). Free carriers' concentration increases with the protonation level from ∼5.5 × 1019 to 2.5 × 1022 cm−3 indicating an increase in delocalized carriers. The figure also demonstrates the sharp increase in the carriers' concentration when protonation increases from QP-PANI to HP-PANI which could explain the metal-insulator boundary conduction transition. Since PANI:CSA is a p-type semiconductor,48 the increase in carriers' concentration to two orders of magnitude (from ∼1019 to 1022 cm−3 when PANI is protonated from QP to FP) suggests that with protonation the Fermi level moves toward the valence band, and this will result in an increase in the WF.
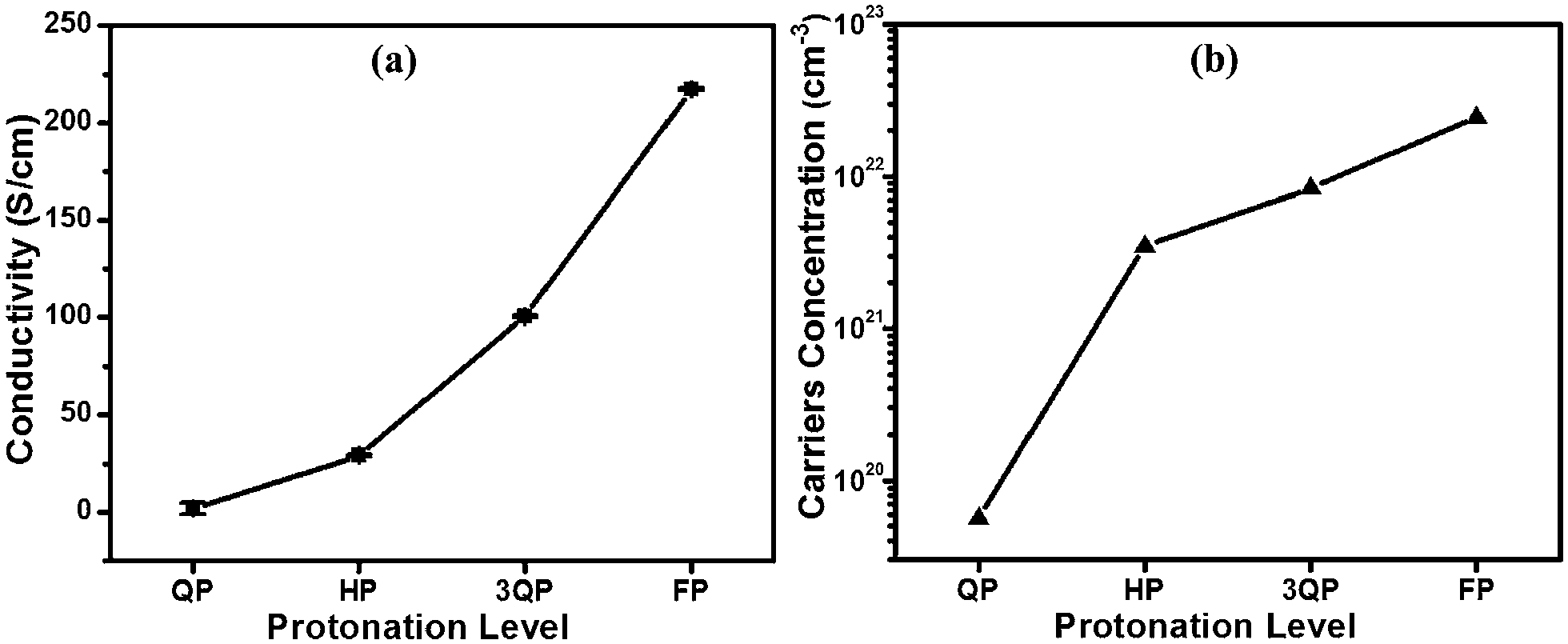 | ||
| Fig. 7 Effect of CSA protonation levels on the electrical conductivity of PANI, and (b) carriers' concentration of PANI:CSA versus protonation levels. | ||
The influence of CSA protonation levels on the WF of PANI was studied using XPS as shown in Fig. 8. In this study, the kinetic energy cutoff was obtained by subtracting the binding energy from the photon energy of Al Kα (1486.6 eV) as shown in Fig. 8(a). The work function was then determined from the intersection of the extrapolated tangent of the cutoff curve with the x-axis at the point where the y-axis equals zero. Fig. 8(b) shows the WF characterization results for the PANI:CSA film surface before etching using three different measurements for each protonation level (i.e., three points each on two films cast from the same solution, as well as a third film cast from another solution prepared by the same procedure) to ensure the repeatability and consistency of the results. The figure shows a wide error bar indicating that the WF of PANI fluctuates. However, the general trend shows an increase in the WF of PANI with increasing CSA level. The work function of PANI measured on the surface of the samples increased from 4.42 ± 0.14 eV for QP-PANI to 4.78 ± 0.13 eV for FP-PANI.
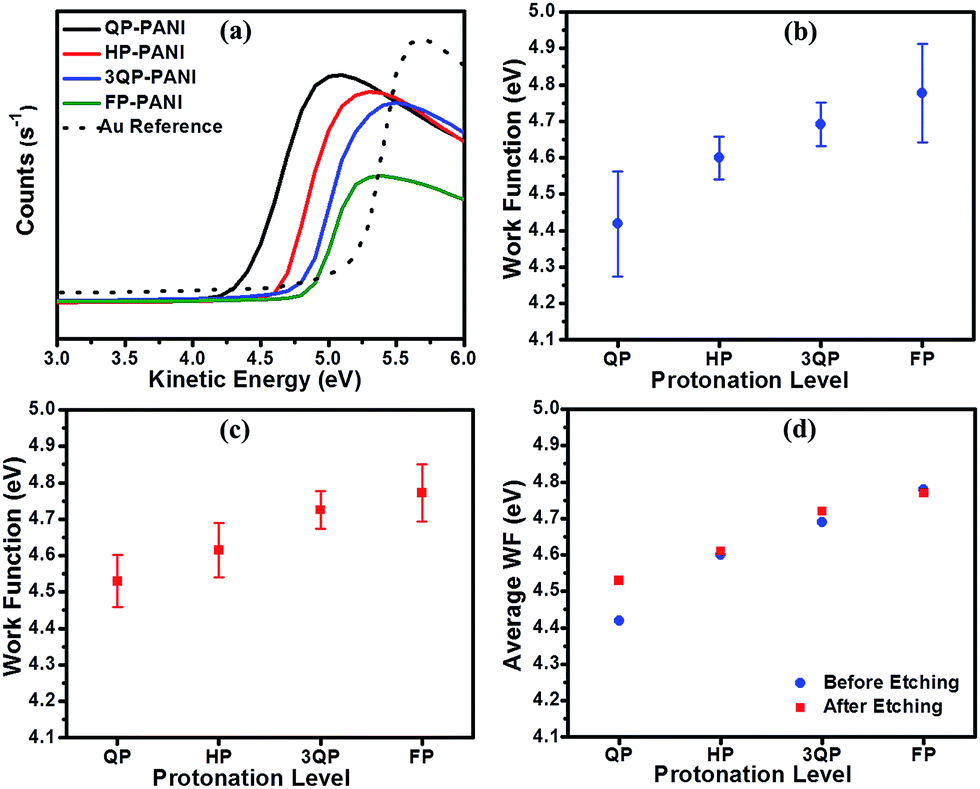 | ||
| Fig. 8 (a) Kinetic energy cut-off of XPS measured on PANI:CSA films, (b) work function of PANI surface before etching, (c) work function of PANI surface after etching, and (d) average work function of PANI before and after etching. | ||
The WF correlation with protonation level is due to the effect of protonation on the internal electronic structure of the PANI. The WF is lowered due to the generation of a large number of free holes (radical cations in the form of bipolarons and polarons) in the polymer chains as it is asserted in Fig. 7(b) presented earlier, resulting in a p-type polymer.48,49 It is well known that during the protonation of PANI, the electronic structure undergoes an internal redox, which forms bipolarons.20 Each bipolaron can then separate to form two polarons, which are generally more stable, especially when they are delocalized over several units of the polymer. These electronic transformations are critical to the evolution of the work function in the PANI:CSA (or other protonic acid-doped PANI) system.
To obtain deeper insight into the effect of surface conditions on the WF, the surface of the various PANI samples was etched using an ion beam in order to remove a few layers from the surface and subsequently determine any fluctuation compared to the initial WF measurements as shown in Fig. 8(c). WF values after etching exhibit a slight decrease over the whole range of protonation. However, the average values of WF before and after ion beam etching presented in Fig. 8(d) demonstrate a similar relationship between WF and protonation level with insignificant change in values, especially at the higher levels of protonation.
Previous publications regarding the work function of polyaniline are limited, and the explanations regarding the change in work function with doping are not explicit in many cases. A summary of these reports is listed in Table 1. The data reported in the literature present various trends when PANI is protonated with various acids as shown in the table. In general, in addition to protonation levels, the WF of PANI depends on various parameters, such as the method used to prepare samples and the procedure followed for protonation, and these factors could account for variations in the WF values and trends reported by various other researchers. However, the investigators who determined absolute values of WF showed a range from ∼4.4 to 5 eV (similar to reported here), which is a suitable range for the recombination layer in organic tandem solar cells.
| Dopant type | PANI:dopant ratio |
Solvent | Conductivity (S cm−1) | WF (eV) | WF trend with increasing dopant | Method of determining WF | Ref. |
|---|---|---|---|---|---|---|---|
| Polystyrene sulfonate | 1:1 to 1:8 |
Water, xylene, or alcohol | 8 × 10−7 to 1 × 10−2 | 4.75 to 5.06 | Increases up to 1:4 then decreases |
Kelvin probe | 21 |
| Sulfuric, trifluoroacetic, perchloric, and hydrochloric acids | Not specified | Formic acid then electrochemical deposition | 0.03 to 0.2 | ΔWF (no absolute value) | No trend presented | Kelvin probe | 28 |
| Triflic acid or tetrafluoroboric | 1:0 to 1:1 |
DMF | No specific conductivity | ΔWF (no absolute value) | Decreases with increasing dopant | Field-effect transistor | 29 |
| Hydrochloric acid | Not specified | Water | Not specified | ΔWF (no absolute value) | Decreases with increasing dopant | Kelvin probe | 30 |
| Camphorsulfonic acid | 1:0.6 to 1:2 |
m-Cresol | 7.5 to 10.5 | 4.7 to 4.8 | No trend with increasing dopant | Kelvin probe | 31 |
| Camphorsulfonic acid | 1:0.25 to 1:1 |
m-Cresol | 2 to 217 | 4.42 ± 0.14 to 4.78 ± 0.13 | Increases with increasing dopant | XPS | Current work |
Conclusions
We have investigated the effect of camphorsulfonic acid protonation levels on the work function of polyaniline films. This study was performed to develop a method to tune the work function of polyaniline for future use as a hole, electron, or recombination interlayer in organic solar cells. Protonation with CSA shows a significant effect on the work function of polyaniline, offering an easy and reliable way to tune the work function of PANI. The work function of PANI exhibits a linear increase with increasing CSA protonation for the levels we investigated. Protonation with CSA produces a p-type polymer that will increase the absolute value of the work function. The results suggest that high protonated polyaniline films are suitable for use as hole transport layers in organic solar cells, while low protonated polyaniline films are suitable for use as electron transport layers in organic solar cells.Acknowledgements
The financial support from National Science Foundation (NSF/EPS-1003970) is greatly appreciated. The editorial assistance of Dr Marinelle Ringer is also acknowledged.Notes and references
- O. Abdulrazzaq, V. Saini, S. Bourdo, E. Dervishi and A. S. Biris, Part. Sci. Technol., 2013, 31(5), 427 CrossRef CAS.
- V. Saini, Z. Li, S. Bourdo, E. Dervishi, Y. Xu, X. Ma, V. P. Kunets, G. J. Salamo, T. Viswanathan, A. R. Biris, D. Saini and A. S. Biris, J. Phys. Chem. C, 2009, 113(19), 8023 CAS.
- A. Petre, S. Diaham, E. Reyes-Melo, V. Saini, L. Zhongrui, E. Dervishi, X. Yang and A. S. Biris, IEEE Trans. Ind. Appl., 2010, 46(2), 627 CrossRef CAS.
- J. Meiss, T. Menke, K. Leo, C. Uhrich, W. Gnehr, S. Sonntag, M. Pfeiffer and M. Riede, Appl. Phys. Lett., 2011, 99, 043301 CrossRef PubMed.
- J. Yun, J. Yeo, J. Kim, H. Jeong, D. Kim, Y. Noh, S. Kim, B. Ku and S. Na, Adv. Mater., 2011, 23(42), 4923 CrossRef CAS PubMed.
- M. Jørgensen, K. Norrman and F. C. Krebs, Sol. Energy Mater. Sol. Cells, 2008, 92(7), 686 CrossRef PubMed.
- B. Ecker, J. C. Nolasco, J. Pallarés, L. F. Marsal, J. Posdorfer, J. Parisi and E. Von Hauff, Adv. Funct. Mater., 2011, 21(14), 2705 CrossRef CAS.
- J. W. Jung, J. U. Lee and W. H. Jo, J. Phys. Chem. C, 2009, 114(1), 633 Search PubMed.
- M. D. Irwin, D. B. Buchholz, A. W. Hains, R. P. H. Chang and T. J. Marks, Proc. Natl. Acad. Sci., 2008, 105(8), 2783 CrossRef CAS.
- K. Fehse, G. Schwartz, K. Walzer and K. Leo, J. Appl. Phys., 2007, 101(12), 124509 CrossRef PubMed.
- O. Abdulrazzaq, S. Bourdo, V. Saini, V. G. Bairi, E. Dervishi, T. Viswanathan, Z. A. Nima and A. S. Biris, Energy Technol., 2013, 1(8), 463 CrossRef CAS.
- Q. T. Le, F. Nüesch, L. J. Rothberg, E. W. Forsythe and Y. Gao, Appl. Phys. Lett., 1999, 75(10), 1357 CrossRef CAS PubMed.
- V. Saini, O. Abdulrazzaq, S. Bourdo, E. Dervishi, A. Petre, V. G. Bairi, T. Mustafa, L. Schnackenberg, T. Viswanathan and A. S. Biris, J. Appl. Phys., 2012, 112(5), 54327–54331 CrossRef PubMed.
- P. Ramidi, O. Abdulrazzaq, C. M. Felton, Y. Gartia, V. Saini, A. S. Biris and A. Ghosh, Energy Technol., 2014, 2(7), 604 CrossRef CAS.
- J. You, C. Chen, Z. Hong, K. Yoshimura, K. Ohya, R. Xu, S. Ye, J. Gao, G. Li and Y. Yang, Adv. Mater., 2013, 25(29), 3973 CrossRef CAS PubMed.
- Y. Zhou, C. Fuentes-Hernandez, J. W. Shim, T. M. Khan and B. Kippelen, Energy Environ. Sci., 2012, 5(12), 9827 CAS.
- T. Ameri, G. Dennler, C. Lungenschmied and C. J. Brabec, Energy Environ. Sci., 2009, 2(4), 347 CAS.
- Y. Yuan, J. Huang and G. Li, Green, 2011, 1, 65 CrossRef CAS.
- A. G. MacDiarmid, Rev. Mod. Phys., 2001, 73, 701 CrossRef CAS.
- D. M. Tigelaar, W. Lee, K. A. Bates, A. Saprigin, V. N. Prigodin, X. Cao, L. A. Nafie, M. S. Platz and A. J. Epstein, Chem. Mater., 2002, 14(3), 1430 CrossRef CAS.
- J. R. Posdorfer, B. Werner, B. Wessling, S. Heun and H. Becker, Proc. SPIE, 2004, 5214, 188 CrossRef CAS PubMed.
- A. G. MacDiarmid and A. J. Epstein, Synth. Met., 1995, 69(1), 85–92 CrossRef CAS.
- E. Gomes and M. Oliveira, Am. J. Polym. Sci., 2012, 2(2), 5 CrossRef CAS PubMed.
- H. Ma, Y. Li, S. Yang, F. Cao, J. Gong and Y. Deng, J. Phys. Chem. C, 2010, 114(20), 9264 CAS.
- T. C. Leung, C. L. Kao, W. S. Su, Y. J. Feng and C. T. Chan, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68(19), 195408 CrossRef.
- R. Schlaf, H. Murata and Z. H. Kafafi, J. Electron Spectrosc. Relat. Phenom., 2001, 120(1–3), 149 CrossRef CAS.
- Y. Wan, Y. Li, Q. Wang, K. Zhang and Y. Wu, The Relationship of Surface Roughness and Work Function of Pure Silver by Numerical Modeling, Int. J. Electrochem. Sci., 2012, 7, 5204–5216 CAS.
- D. Chinn, J. DuBow, M. Liess, M. Josowicz and J. Janata, Chem. Mater., 1995, 7(8), 1504 CrossRef CAS.
- B. J. Polk, K. Potje-Kamloth, M. Josowicz and J. Janata, J. Phys. Chem. B, 2002, 106(44), 11457 CrossRef CAS.
- P. Jinsung, B. Doyeon, J. Kuewhan, H. Seungjoo, Y. Jaemoon and N. Sungsoo, Nanotechnology, 2012, 23(36), 365705 CrossRef PubMed.
- H. A. Ahmad, D. Nayak and S. Panda, J. Appl. Polym. Sci., 2013, 129(1), 230 CrossRef CAS.
- O. I. Klyushnikov, J. Struct. Chem., 1998, 39(6), 944 CrossRef CAS.
- M. Trchová and J. Stejskal, Pure Appl. Chem., 2011, 83(10), 1803 CrossRef.
- C. J. Mathai, S. Saravanan, M. R. Anantharaman, S. Venkitachalam and S. Jayalekshmi, J. Phys. D: Appl. Phys., 2002, 35(17), 2206 CrossRef CAS.
- M. Babazadeh, J. Appl. Polym. Sci., 2009, 113(6), 3980 CrossRef CAS.
- A. P. Monkman, G. C. Stevens and D. Bloor, J. Phys. D: Appl. Phys., 1991, 24(5), 738 CrossRef CAS.
- R. S. Biscaro, M. C. Rezende and R. Faez, Polym. Adv. Technol., 2009, 20(1), 28 CrossRef CAS.
- A. Lodha, S. M. Kilbey, P. C. Ramamurthy and R. V. Gregory, J. Appl. Polym. Sci., 2001, 82(14), 3602 CrossRef CAS.
- Y. Wei and K. F. Hsueh, J. Polym. Sci., Part A: Polym. Chem., 1989, 27(13), 4351 CrossRef CAS.
- Y. Xia, A. G. MacDiarmid and A. J. Epstein, Macromolecules, 1994, 27(24), 7212 CrossRef CAS.
- J. P. Pouget, M. E. Jozefowicz, A. J. Epstein, X. Tang and A. G. MacDiarmid, Macromolecules, 1991, 24(3), 779 CrossRef CAS.
- X. Zhang, J. Zhu, N. Haldolaarachchige, J. Ryu, D. P. Young, S. Wei and Z. Guo, Polymer, 2012, 53(10), 2109 CrossRef CAS PubMed.
- K. Lee, S. Cho, S. Heum Park, A. J. Heeger, C. Lee and S. Lee, Nature, 2006, 441(7089), 65 CrossRef CAS PubMed.
- S. Saravanan, C. Joseph Mathai, M. R. Anantharaman, S. Venkatachalam and P. V. Prabhakaran, J. Phys. Chem. Solids, 2006, 67(7), 1496 CrossRef CAS PubMed.
- M. Khalid, M. A. Tumelero, I. S. Brandt, V. C. Zoldan, J. Acu and A. A. Pasa, Indian Journal of Materials Science, 2013, 2013, 7 CrossRef PubMed.
- W. Łużny and E. Bańka, Macromolecules, 2000, 33(2), 425 CrossRef.
- E. R. Holland, S. J. Pomfret, P. N. Adams and A. P. Monkman, J. Phys.: Condens. Matter, 1996, 8(17), 2991 CrossRef CAS.
- H. Anno, M. Hokazono, F. Akagi, M. Hojo and N. Toshima, J. Electron. Mater., 2013, 42(7), 1346 CrossRef CAS.
- K. Mamma, K. Siraj and N. Meka, Am. J. Polym. Sci. Eng., 2013, 1(1), 1 Search PubMed.
| This journal is © The Royal Society of Chemistry 2015 |