
Stability indicating RP-HPLC method for the determination of flubendazole in pharmaceutical dosage forms
Nada S. Abdelwahab and
Maha M. Abdelrahman*
Pharmaceutical Analytical Chemistry Department, Faculty of Pharmacy, Beni-Suef University, Alshaheed Shehata Ahmad Hegazy St., 62514, Beni-Suef, Egypt. E-mail: nadasayed2003@yahoo.com; Fax: +2-0282317950; Tel: +2-01117236884
First published on 15th December 2014
Abstract
This paper presents a stability study of the drug flubendazole (FLUB) – a benzimidazole carbamate derivative with anthelmintic activity. In order to investigate the stability of the drug, FLUB was subjected to stress degradation under different conditions, as recommended by the International Conference on Harmonization (ICH). A validated HPLC-photodiode array detector (DAD) method was established to resolve FLUB from all its degradation products obtained under acidic, basic, neutral, oxidation, photo-degradation, and thermal conditions, and also from its suspension additives. Chromatographic separation was achieved on a ZORBAX Eclipse Plus C18 column in an isocratic mode of elution using a mixture of water![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :acetonitrile (50:50, v/v) as a mobile phase, at a flow rate of 1 mL min−1, where FLUB was well resolved from all its degradation products at a tR of 5.35 min. The thermostated column compartment (TCC) was adjusted to 25 °C and the effluent was monitored by a photo-diode array detector at 254 nm, and FLUB was found to be linear in the range of 0.5–10 μg mL−1. The method was validated in terms of accuracy, precision, specificity, robustness, and ruggedness, as per USP guidelines. It was successfully applied to quantify FLUB in bulk powder and in pharmaceutical formulations with complete resolution between FLUB and its suspension additives. Statistical analyses between the suggested method and the official HPLC method using student's-t and F-ratio tests reveal that the suggested method is as accurate and precise as the reported one.
:acetonitrile (50:50, v/v) as a mobile phase, at a flow rate of 1 mL min−1, where FLUB was well resolved from all its degradation products at a tR of 5.35 min. The thermostated column compartment (TCC) was adjusted to 25 °C and the effluent was monitored by a photo-diode array detector at 254 nm, and FLUB was found to be linear in the range of 0.5–10 μg mL−1. The method was validated in terms of accuracy, precision, specificity, robustness, and ruggedness, as per USP guidelines. It was successfully applied to quantify FLUB in bulk powder and in pharmaceutical formulations with complete resolution between FLUB and its suspension additives. Statistical analyses between the suggested method and the official HPLC method using student's-t and F-ratio tests reveal that the suggested method is as accurate and precise as the reported one.
1. Introduction
Flubendazole (FLUB) is chemically is identified as [5-(4-fluorobenzoyl)-1H-benzimidazol-2-yl]carbamic acid methyl ester.1 It is a benzimidazole carbamate derivative, with an anthelmintic effect and activity against most nematodes and some other worms. Its positive activity against some larval stages and ova has also been demonstrated. It works by inhibiting or destroying cytoplasmic microtubules in the worm's intestinal or absorptive cells, leading to the inhibition of glucose uptake and depletion of the glycogen stores, and hence leads to the death of the worm within several days.2A thorough literature survey has revealed the presence of few methods for the determination of FLUB. European Pharmacopoeia3 has reported a RP-HPLC assay of FLUB. Also, different HPLC-MS methods have been described for the determination of FLUB in pig4 and in eggs and poultry muscle,5 with its metabolites, or in environmental6 and wastewater7 samples. The drug has also been analyzed, along with its metabolites, in plasma by UV photodiode-array and fluorescence detection.8 Additionally, FLUB and other benzimidazole compounds have been resolved by HPLC methods in bovine liver9 and in milk samples.10 Moreover, it has also been determined by different polarographic methods in either pharmaceutical formulations11 or in biological fluids.12 Furthermore, an HPLC-UV method for the simultaneous determination of FLUB and febantel in swine and poultry feed has been published.13 However, until now, no stability indicating method for the determination of FLUB in the presence of its degradation products or suspension additives has been previously established.
Current state-of-the-art argues that stability is the most important quality requirement for a pharmaceutical product. Stable preparations directly emphasize the quality of the product, assuring its precise delivery. Moreover, the drug product shelf-life depends on the analytical studies performed under normal and stressed conditions.14 The present drug stability test guidelines, Q1A (R2),15,16 issued by the International Conference on Harmonization (ICH) recommend carrying out stress testing on drug substances to establish their inherent stability characteristics. The analysis of stability samples should be performed through the use of validated stability-indicating analytical methods (SIAM).17 It is essential for SIAM procedures to be able to accurately measure the active ingredients, without interference, from the degradation products, excipients, impurities, or other potential process contaminations.18,19
As previously mentioned, a comprehensive HPLC study of the degradation behavior of FLUB under various ICH prescribed stress conditions has been lacking to date. Accordingly, the present paper assesses the degradation behavior of FLUB under acidic, basic, neutral, oxidation, photo-degradation, and thermal conditions, and develops a RP-HPLC-DAD method to separate the drug and its degradation products in a reasonable retention time, which was successfully applied for the determination of FLUB in pharmaceutical dosage forms.
2. Experimental
2.1. Instrument
HPLC measurements were carried out on an Agilent 1260 Infinity instrument, Germany, equipped with an Agilent 1260 Infinity preparative pump (G1361A), Agilent 1260 Infinity Diode array detector VL (G131SD), Agilent 1260 Infinity Thermostated column compartment (G1316A) and an Agilent 1260 Infinity preparative Autosampler (G2260A). Separation and quantitation were performed on a ZORBAX Eclipse Plus C18 column (USA) (250 mm × 4.6 mm i.d., 5 μm particle size).2.2. Materials
(b) Fluver® tablets (batch no. 3139001) and suspension (batch no. 3209033) were manufactured by Alexandria Co. for Pharmaceuticals & Chemical Industries, Alexandria, Egypt. It was claimed that the content was 100 mg of FLUB per tablet or 5 mL in suspension.
(b) Deionized water was obtained from SEDICO pharmaceuticals Co., Cairo, Egypt.
(c) Hydrochloric acid, sodium hydroxide, and hydrogen peroxide (analytical grade) were purchased from El-Nasr Pharmaceutical Chemicals Co., Abu-Zabaal, Cairo, Egypt.
2.3. Standard solutions
(a) A stock standard solution of FLUB (1 mg mL−1) was prepared by accurately weighing 0.1 g FLUB in a 100 mL volumetric flask and dissolving in 0.1 M methanolic HCl.(b) A working standard solution of FLUB (0.1 mg mL−1) was prepared by a suitable dilution of the FLUB stock solution with water:acetonitrile (50:50, v/v).
3. Methods
3.1. Chromatographic conditions
Chromatographic separation was achieved on a ZORBAX Eclipse Plus C18 column (250 mm × 4.6 mm i.d., 5 μm particle size) using a mobile phase consisting of a mixture of water and acetonitrile (50:50, v/v) in isocratic mode. Separation was performed at 25 °C using a 1 mL min−1 flow rate and a run time of 8 min. The injection volume was 50 μL and the photodiode array detector was adjusted at 230 nm and 254 nm.
3.2. Linearity and construction of the calibration curve
To establish the linearity of the analytical method, a series of dilutions ranging from 0.5–10 μg mL−1 of pure FLUB were prepared from the working standard solution (0.1 mg mL−1) using a mixture of water:acetonitrile (50:50, v/v) as a solvent. Triplicate 50 μL injections were made for each concentration and were chromatographed under the previously mentioned chromatographic conditions. The relative peak areas at 254 nm (using 5 μg mL−1 of FLUB as an external standard) were plotted against the corresponding concentrations to obtain the calibration graph.
3.3. Stability study of flubendazole
A flubendazole forced-degradation study was carried out under the range of conditions recommended in the ICH guidelines, such as acidic, basic, neutral, oxidation, photo-degradation, and thermal conditions, in the dark for all conditions except for photo-degradation – in order to exclude the possible degradation effect of light on FLUB. A stock standard solution of FLUB (1 mg mL−1) was used throughout the stress decomposition study to give an indication of the stability indicating property and the specificity of the proposed method. Using the previously computed regression equation, the remaining FLUB concentration in each sample was determined and the % FLUB degradation was calculated.:water (50:50, v/v) to obtain a sample solution of 200 μg mL−1. A concentration of 10 μg mL−1 was then prepared, from which 50 μL was injected into the LC system.3.4. Application to pharmaceutical formulations
- Fluvermal® and Fluver® tablets: ten tablets each of Fluvermal® and Fluver® tablets were separately weighed, powdered, and well mixed. An amount of powdered tablets from each dosage form equivalent to 100 mg of FLUB was taken and dissolved in 75 mL of 0.1 M methanolic HCl for 15 min; the volume was then adjusted to 100 mL with 0.1 M methanolic HCl and then filtered. A suitable dilution of the prepared solutions with water:acetonitrile (50:50, v/v) was performed to obtain a working solution (0.1 mg mL−1) of each pharmaceutical formulation.
- Fluvermal® and Fluver® suspensions: 5 mL from each suspension was accurately transferred into two separate 100 mL volumetric flasks. 75 mL of 0.1 M methanolic HCl was added and ultra-sonicated for 10 min; then the volume was completed to the mark with 0.1 M methanolic HCl and then filtered to obtain a stock solution of 1 mg mL−1 FLUB. Working solutions of 0.1 mg mL−1 were then prepared in a water:acetonitrile (50:50, v/v) mixture.
- Different concentrations of each formulation were prepared and then the procedure was followed under chromatographic conditions. In order to assess the accuracy of the method, recovery studies were performed by spiking the pre-analyzed FLUB sample (4 μg mL−1) with an extra 80%, 100%, and 120% of pure FLUB; then, the mean % recovery of the pure added drug was calculated.
4. Results and discussion
According to the International Conference on Harmonization (ICH) guidelines, the requirements for establishing SIAMs are now mandatory.15,16 The purpose of stability testing is to provide evidence on how the quality of a drug substance or drug product varies with time under the influence of a variety of environmental factors, such as temperature, light, oxygen, pH, and moisture. In this work, a stability study of FLUB was carried out for the first time using an HPLC-DAD method. The developed method was able to resolve the drug from all its degradation products in a single run; moreover, the drug was completely resolved from the suspension excipients. Additionally, the stress degradation conditions were followed for the FLUB pharmaceutical tablets and the same results were obtained as for the pure standard drug.4.1. Method development and optimization
Many parameters must be evaluated and optimized during the method development and optimization in order to separate the parent drug from the suspension additives and degradation products within a single run without any interference.In order to select a suitable mobile phase for the analysis of FLUB, the pure drug, along with its degradation products and pharmaceutical formulations, were injected and run in different solvent systems on the basis of trial and error, taking into consideration the system suitability parameters, retention time, tailing factor, the number of theoretical plates, and HETP. Initially methanol and water in different ratios were tried. However, the pure drug started to be eluted very late (after more than 20 min), therefore, methanol was replaced by acetonitrile. A water:acetonitrile mixture was used in different ratios and it was found that using a mixture of water and acetonitrile (50:50, v/v) as a mobile phase at a flow rate of 1 mL min−1 gave an acceptable retention time (tR), theoretical plates, and a good resolution between the drug, the suspension excipients, and the degradation products.
Different stationary phases were tested, such as ZORBAX Eclipse Plus C18 and C8 columns. The two stationary phases nearly had the same the system suitability parameters.
In addition, the mobile phase was delivered at different flow rates (0.8 mL min−1, 1 mL min−1, and 1.5 mL min−1) in order to obtain the maximum resolution within the shortest analysis time. Pumping the mobile phase at 1 mL min−1 resulted in a good resolution within 8 min analysis time.
The photodiode array detector (DAD) was adjusted at different detection wavelengths (230 nm and 254 nm) in order to detect any degradation products produced in any forced-degradation conditions and for increasing method sensitivity. It was found that the number of detected degradation products was the same at both wavelengths, but scanning at 254 nm gave a higher sensitivity for FLUB than scanning at 230 nm.
The thermostated column compartment temperature was optimized by testing different temperatures (20 °C, 25 °C, and 30 °C). It was observed that the column temperature neither affected the chromatographic separation nor the peak shape.
Finally, the chromatographic separation of FLUB, its degradation products, and suspension excipients was carried out on a ZORBAX Eclipse Plus C18 column with a mixture of water:acetonitrile (50:50, v/v) delivered at 1 mL min−1, maintaining the column temperature at 25 °C and the detection at 254 nm.
4.2. Results of the stress degradation studies
A summary of the results of the FLUB stability studies is given in Table 1.| Stress conditions | Time of degradation (h) | tR of the obtained degradation products (min) | % Degradation |
|---|---|---|---|
| 0.1 N NaOH at 80 °C | 5 | 2.52 | 22.5% |
| 1 N NaOH at 80 °C | 5 | 2.22, 2.7, and 2.8 | 99.25% |
| 0.1 N HCl at 80 °C | 5 | No degradation | 0% |
| 1 N HCl at 80 °C | 5 | 2.21 and 2.47 | 33.10% |
| H2O at 80 °C | 5 | No degradation | 0% |
| 3% H2O2 at 80 °C | 5 | 2.47 and 3.23 | 17.9% |
| 30% H2O2 at room temperature | 5 | 2.49 and 3.20 | 43.66% |
| Photo-degradation | 20 | 3.15, 3.52, and 3.78 | 8.50% |
| Dry heat at 80 °C | 5 | No degradation | 0% |
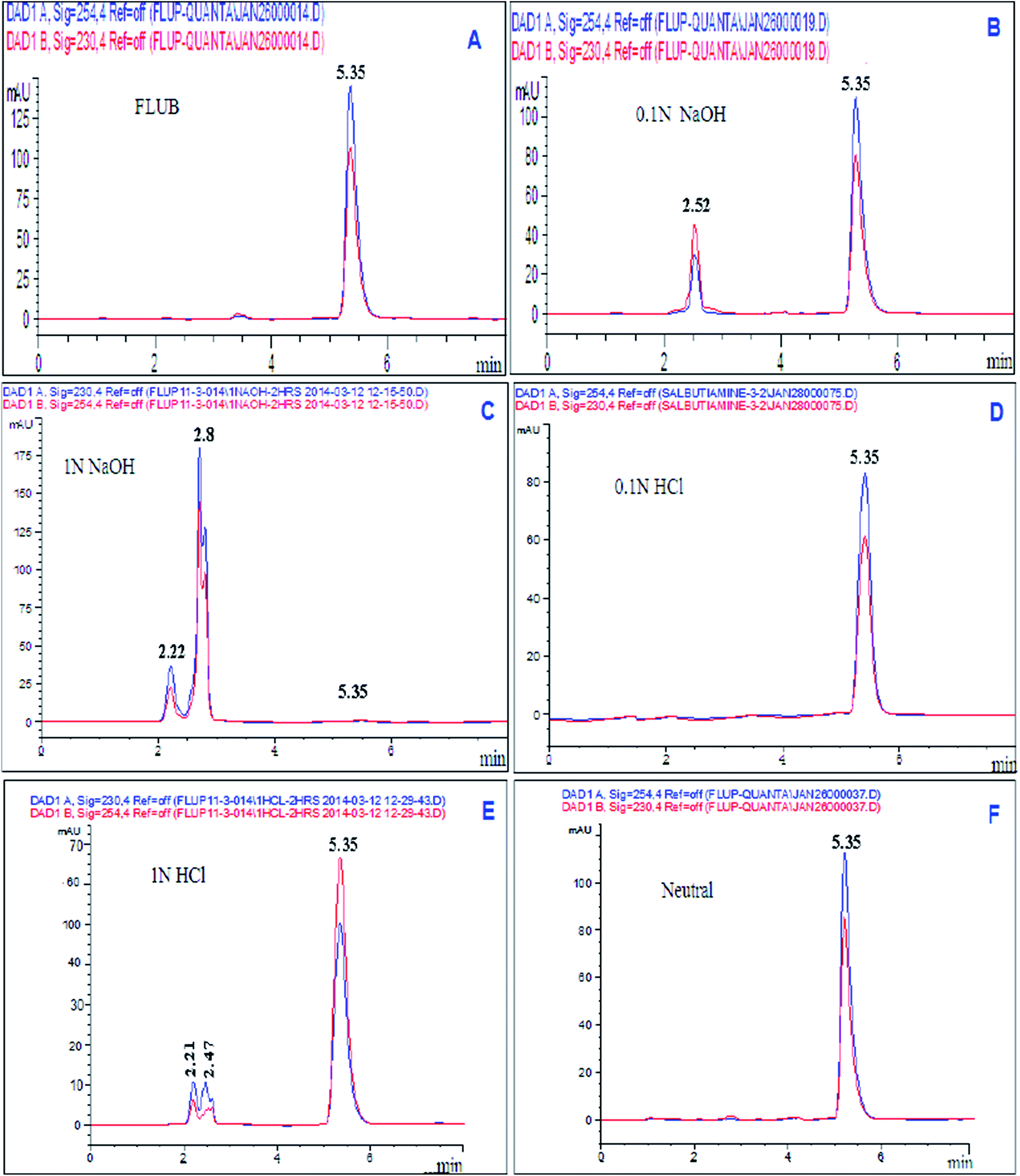 | ||
| Fig. 1 HPLC chromatogram of (A) 10 μg mL−1 of pure FLUB, (B) alkaline degradation using 0.1 N NaOH, (C) alkaline degradation using 1 N NaOH, (D) acidic degradation using 0.1 N HCl, (E) acidic degradation using 1 N HCl, and (F) neutral degradation under neutral conditions. | ||
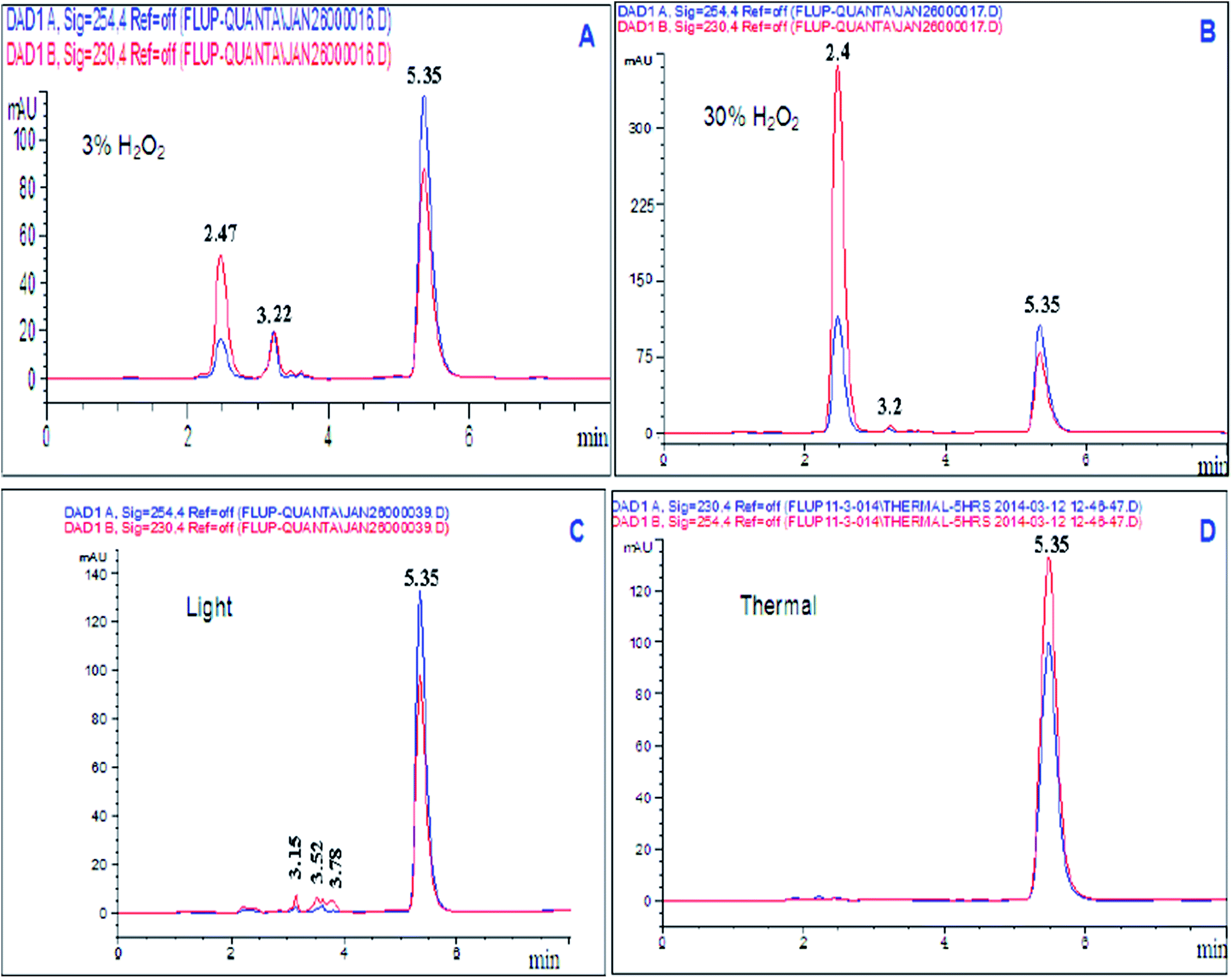 | ||
| Fig. 2 HPLC chromatogram of (A) degradation using 3% H2O2, (B) degradation using 30% H2O2, (C) photo-degradation under light conditions, and (D) degradation under thermal conditions. | ||
4.3. Application of the method
The second step after method optimization was its application to determine FLUB in its pure form and in different pharmaceutical formulations. The method showed good linearity when the relative peak area (using 5 μg mL−1 of FLUB as an external standard) was plotted against FLUB concentration in the range of 0.5–10 μg mL−1, and the computed regression equation was found to be:| A = 0.1944C + 0.0197, r = 0.9998 |
| Parameters | FLUB |
|---|---|
| a The intraday (n = 9) average of three different concentrations (4 μg mL−1, 8 μg mL−1, and 10 μg mL−1) repeated three times within a single day.b The interday (n = 9) average of three different concentrations (4 μg mL−1, 8 μg mL−1, and 10 μg mL−1) repeated three times in three successive days. | |
| Linearity | |
| Range (μg mL−1) | 0.5–10 |
| Slope | 0.1944 |
| SE of the slope | 0.0079 |
| Intercept | 0.0197 |
| SE of the intercept | 0.0013 |
| Correlation coefficient | 0.9998 |
| Accuracy (mean ± % RSD) | 100.57 ± 1.47 |
|
|
| Precision (% RSD) | |
| Repeatabilitya | 1.01 |
| Intermediate precisionb | 1.02 |
| LOD (μg mL−1) | 0.11 |
| LOQ (μg mL−1) | 0.34 |
In order to test the ability of the method to quantify FLUB in its marketed formulations, it was applied to determine FLUB in Fluvermal® and Fluver® tablets and suspensions. Good percentage recoveries were obtained, as given in Tables 3 and 4; also the chromatograms in Fig. 3 showed a complete resolution between the peak of the parent drug from all the peaks of the suspension additives. Also, the results of the recovery studies were found to be acceptable at all tested levels, proving the high accuracy of the proposed method (see Tables 3 and 4).
| Pharmaceutical formulation | Taken | Found | % Founda ± % RSD | Standard addition technique | |
|---|---|---|---|---|---|
| Pure added (μg mL−1) | % Foundb | ||||
| a Average of 6 determinations.b Average of 3 determinations. | |||||
| Fluvermal® tablets (B. N. DCE1053) claimed to contain 100 mg FLUB/tablet | 4.00 | 4.03 | 100.75 ± 1.763 | 4.00 | 101.00 |
| 5.00 | 99.20 | ||||
| 6.00 | 97.33 | ||||
| Mean ± % RSD | 99.18 ± 1.85 | ||||
| Fluvermal® suspension (B. N. DFE1824) claimed to contain 100 mg FLUB/t5 mL | 4.00 | 4.26 | 106.50 ± 0.133 | 4.00 | 100.25 |
| 5.00 | 100.60 | ||||
| 6.00 | 97.50 | ||||
| Mean ± % RSD | 99.45 ± 1.71 | ||||
| Pharmaceutical formulation | Taken | Found | % Founda ± % RSD | Standard addition technique | |
|---|---|---|---|---|---|
| Pure added (μg mL−1) | % Foundb | ||||
| a Average of 6 determinations.b Average of 3 determinations. | |||||
| Fluver® tablets (B. N. 3139001) claimed to contain 100 mg FLUB/tablet | 4.00 | 4.01 | 100.25 ± 0.439 | 4.00 | 100.00 |
| 5.00 | 103.40 | ||||
| 6.00 | 101.83 | ||||
| Mean ± % RSD | 101.74 ± 1.67 | ||||
| Fluver® suspension (B. N. 3209033) claimed to contain 100 mg FLUB/5 mL | 4.00 | 4.17 | 104.25 ± 1.322 | 4.00 | 99.25 |
| 5.00 | 97.40 | ||||
| 6.00 | 98.33 | ||||
| Mean ± % RSD | 98.33 ± 1.33 | ||||
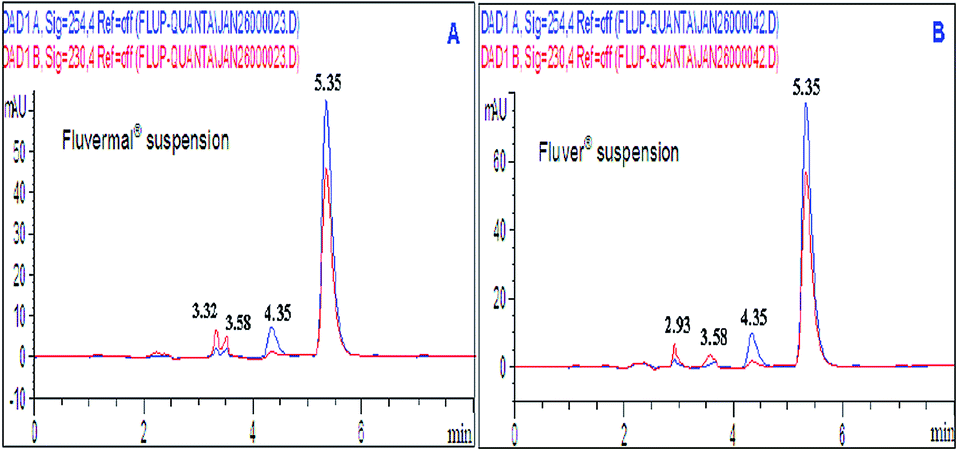 | ||
| Fig. 3 HPLC chromatogram of (A) Fluvermal® suspension and (B) Fluver® suspension using the proposed LC method. | ||
The results obtained by applying the proposed HPLC-DAD method for the determination of FLUB in bulk powder was statistically compared with those obtained by the official HPLC method3 using student's t-test and variance ratio F-test, revealing no significant difference between the performance of the two methods regarding accuracy and precision (see Table 5).
| Items | HPLC method | Official methoda [ref. 3] |
|---|---|---|
| a RP-HPLC using a gradient mixture of ammonium acetate:acetonitrile at a flow rate of 1.2 mL min−1 and UV detection at 250 nm.b The figures between the parenthesis represent the corresponding tabulated values of t and F at P = 0.05. |
||
| Mean | 100.56 | 100.22 |
| % RSD | 1.47 | 1.27 |
| Variance | 2.19 | 1.62 |
| n | 9 | 8 |
| Student's t-test | 0.52 (2.13)b | |
| F-test | 1.36 (3.73)b | |
4.4. System suitability testing
The system suitability parameters were calculated to ascertain the suitability and effectiveness of the operating system. These were evaluated by comparing the obtained parameter values given in Table 6, with the acceptance criteria of the FDA guidance document.20 It was found that the capacity factors were within the range of 0.8 < k′ > 10, the resolution (Rs) between two adjacent peaks was greater than 2, the column efficiency (number of theoretical plates, N) was >2000, and the tailing factor was 1.02 (see Table 6).| Parameters | Obtained value | Reference value20 | |
|---|---|---|---|
| FLUB | |||
| a HETP = height equivalent to theoretical plate, cm per plate. | |||
| Tailing factor (T) | 1.02 | ≈1 | |
| Capacity factor (k′) | 3.86 | 1–10 acceptable | |
| Number of theoretical plates (N) | 2488.36 | Increase with efficiency of the separation | |
| HETP | 0.01005 | The smaller the value the higher the column efficiency | |
| Resolution (Rs) | 0.1 N NaOH | 9.91 | R > 2 |
| 1 N NaOH | 7.52 | ||
| 1 N HCl | 8.08 | ||
| 3% H2O2 | 7.03 | ||
| 30% H2O2 | 8.64 | ||
| Neutral | 9.65 | ||
| Light | 5.53 | ||
| Fluvermal® suspension | 2.95 | ||
| Fluver® suspension | 2.95 | ||
| Selectivity (α) | 0.1 N NaOH | 2.99 | >1 |
| 1 N NaOH | 2.50 | ||
| 1 N HCl | 3.10 | ||
| 3% H2O2 | 2.01 | ||
| 30% H2O2 | 2.02 | ||
| Neutral | 3.63 | ||
| Light | 1.16 | ||
| Fluvermal® suspension | 1.31 | ||
| Fluver® suspension | 1.31 | ||
4.5. Method validation
Method validation was performed as recommended in USP.215. Conclusion
In this paper, drug stability guidelines issued by the International Conference on Harmonization (ICH) were followed to establish the inherent stability of FLUB under various different stress conditions, such as, acidic, alkaline, neutral, oxidative, photo-degradation, and thermal degradation conditions. This stability study was performed by a sensitive, specific, accurate, and validated stability indicating HPLC-DAD method. The drug was found to be liable to alkaline and oxidative conditions, and the degradation products were well separated from the drug substance, demonstrating the stability indicating power of the method. Moreover, the method was capable of resolving the suspension formulation additives from the parent drug when applied to real marketed samples. The information presented herein could be very useful for the quality monitoring of FLUB bulk samples and could also be employed to check the quality of the drug during stability studies. The developed LC method fulfilled all the requirements necessary to be identified as a reliable and feasible method, including accuracy, linearity, recovery, and precision data.References
- S. Budavaried, The Merck Index, An Encyclopedia of Chemicals, Drugs and Biologicals, Merck & Co., Inc., 13th edn, 2002 Search PubMed.
- S. C. Sweetman, Martindale: The Complete Drug Reference, The Pharmaceutical Press, London, 36th edn, 2009 Search PubMed.
- European Pharmacopoeia 7.0, 2008, p. 2024.
- M. Nobilis, T. Jira, M. Lísa, M. Holčapek, B. Szotáková, J. Lamka and L. Skálová, Achiral and chiral high-performance liquid chromatographic determination of flubendazole and its metabolites in biomatrices using UV photodiode-array and mass spectrometric detection, J. Chromatogr. A, 2007, 1149, 112–120 CrossRef CAS PubMed.
- H. De Ruyck, E. Daeseleire, K. Grijspeerdt, H. De Ridder, R. Van Renterghem and G. Huyghebaert, Determination of Flubendazole and Its Metabolites in Eggs and Poultry Muscle with Liquid Chromatography-Tandem Mass Spectrometry, J. Agric. Food Chem., 2001, 49, 610–617 CrossRef CAS PubMed.
- J. C. Van De Steene, K. A. Mortier and W. E. Lambert, Tackling matrix effects during development of a liquid chromatographic-electrospray ionisation tandem mass spectrometric analysis of nine basic pharmaceuticals in aqueous environmental samples, J. Chromatogr. A, 2006, 1123, 71–81 CrossRef CAS PubMed.
- J. C. Van De Steene and W. E. Lambert, Validation of a solid-phase extraction and liquid chromatography-electrospray tandem mass spectrometric method for the determination of nine basic pharmaceuticals in wastewater and surface water samples, J. Chromatogr. A, 2008, 1182, 153–160 CrossRef CAS PubMed.
- M. Nobilis, Z. Vybíralová, V. Krízová, V. Kubícek, M. Soukupová, J. Lamka, B. Szotáková and L. Skálová, Sensitive chiral high-performance liquid chromatographic determination of anthelmintic flubendazole and its phase I metabolites in blood plasma using UV photodiode-array and fluorescence detection application to pharmacokinetic studies in sheep, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2009, 876, 89–96 CrossRef PubMed.
- G. Caprioli, G. Cristalli, R. Galarini, D. Giacobbe, M. Ricciutelli, S. Vittori, Y. Zuo and G. Sagratini, Comparison of two different isolation methods of benzimidazoles and their metabolites in the bovine liver by solid-phase extraction and liquid chromatography-diode array detection, J. Chromatogr. A, 2010, 1217, 1779–1785 CrossRef CAS PubMed.
- A. Martínez-Villalba, E. Moyano and M. T. Galceran, Ultra-high performance liquid chromatography-atmospheric pressure chemical ionization-tandem mass spectrometry for the analysis of benzimidazole compounds in milk samples, J. Chromatogr. A, 2013, 1313, 119–131 CrossRef PubMed.
- P. Gratteri, S. Pinzauti, E. La Porta, P. Mura, G. Papeschi and G. Santoni, Determination of flubendazole in pharmaceutical dosage forms by differential pulse polarography and UV spectroscopy, IL Farmaco, 1990, 45, 707–714 CAS.
- N. El-Enany, F. Belal and M. Rizk, Polarographic determination of flubendazole in spiked human urine and plasma, IL Farmaco, 2003, 58, 613–617 CrossRef CAS.
- G. Dusi, V. Gamba and E. Faggionato, Rapid determination of the antiparasitic drugs flubendazole and febantel in feeds by HPLC with ultraviolet detection, J. Pharm. Biomed. Anal., 2005, 38, 375–379 CrossRef CAS PubMed.
- P. Hamrapurkar, P. Patil and S. Pawar, Stress degradation studies and development of a validated stability-indicating-assay-method for determination of diacerein in presence of degradation products, Pharm. Methods, 2011, 2, 30–35 CrossRef PubMed.
- ICH Stability Testing of New Drug Substances and Products Q1A (R2) International Conference on Harmonization, IFPMA, Geneva, 2003.
- ICH Stability Testing: Photostability Testing New Drug Substances and Products (1996) International Conference on Harmonization, IFPMA, Geneva.
- S. Singh and M. Bakshi, Guidance on conduct of stress test to determine inherent stability of drugs, Pharm. Tech. On-line, 2000, 24, 1–14 CrossRef.
- Analytical Procedures and Method Validation Chemistry, Guidance for Industry, S.I.C. Manufacturing and Controls Documentation, US Department of Health and Human Services, FDA, 2000.
- S. Vidovic, B. Stojanovic, J. Veljkovic, L. Prazic-Arsic and G. Roglic, Simultaneous Determination of some Water-Soluble Vitamins and Preservatives in Multivitamin Syrup by Validated Stability-Indicating High-Performance Liquid Chromatography Method, J. Chromatogr. A, 2008, 1202, 155–162 CrossRef CAS PubMed.
- Center for Drug Evaluation and Research, U.S. Food and Drug Administration, Reviewer Guidance, Validation of Chromatographic Methods, FDA, Rockville, MD, November, 1994 Search PubMed.
- The United States Pharmacopeia, National Formulary 35, United States Pharmacopeia Convention Inc., 30th edn, 2012 Search PubMed.
- S. Siddiqui, S. Paliwal, K. Kohli, A. A. Siddiqui and K. Sahu, RP-HPLC Method for Estimation and Stress Degradation Study of Paclitaxel as per ICH Guidelines, J. Chromatogr. Sep. Tech., 2012, 2, 3–4 Search PubMed.
| This journal is © The Royal Society of Chemistry 2015 |