
Typical and atypical domain combinations in human protein kinases: functions, disease causing mutations and conservation in other primates
Ramaswamy Rakshambikai†
,
Malini Manoharan†,
Mutharasu Gnanavel‡
and
Narayanaswamy Srinivasan*
Molecular Biophysics Unit, Indian Institute of Science, Bangalore 560012, India. E-mail: ns@mbu.iisc.ernet.in; Fax: +91-80-2293-2837; Tel: +91-80-2360-0535
First published on 2nd March 2015
Abstract
Ser/Thr and Tyr protein kinases orchestrate many signalling pathways and hence loss in this balance leads to many disease phenotypes. Due to their high abundance, diversity and importance, efforts have been made in the past to classify kinases and annotate their functions at both gross and fine levels. These kinases are conventionally classified into subfamilies based on the sequences of catalytic domains. Usually the domain architecture of a full-length kinase is consistent with the subfamily classification made based on the sequence of kinase domain. Important contributions of modular domains to the overall function of the kinase are well known. Recently occurrence of two kinds of outlier kinases-“Hybrid” and “Rogue” has been reported. These show considerable deviations in their domain architectures from the typical domain architecture known for the classical kinase subfamilies. This article provides an overview of the different subfamilies of human kinases and the role of non-kinase domains in functions and diseases. Importantly this article provides analysis of hybrid and rogue kinases encoded in the human genome and highlights their conservation in closely related primate species. These kinases are examples of elegant rewiring to bring about subtle functional differences compared to canonical variants.
Introduction
Analysis of genomic data shows that more than 70% of eukaryotic proteins are composed of multiple domains.1 As domains determine the function and influence recognition of evolutionary relationships among proteins, it is important to understand the role of domain combinations and their interactions.2 A major proportion of protein kinases focused in this article are multi-domain in nature. Protein kinases that constitute about 2% of the proteomes of most eukaryotes, phosphorylate more than 30% of the cellular proteins.3 Protein phosphorylation and dephosphorylation are the most prevalent modifications regulating the structures and functions of proteins in a wide spectrum of cellular processes, ranging from cell fate control to regulation of metabolism. Therefore a tight regulation of these kinases is required for the normal functioning of any living organism. In humans, aberrant kinase activity is implicated in a variety of diseases, in particular those involving inflammatory or proliferative responses, such as cancer, rheumatoid arthritis, cardiovascular and neurological disorders, asthma and psoriasis.4 Directly or indirectly, more than 400 human diseases have been connected to protein kinases and they are now considered to be the second most important group of drug targets after G-protein-coupled receptors.5 Classical protein kinases have a canonical catalytic domain of about 250 amino acids in length, which consists of a small N-terminal lobe made predominantly of β-sheets and a larger C-terminal lobe rich in α-helical regions. ATP binds in a cleft between the two lobes in such a way that the adenosine moiety is buried in a hydrophobic pocket with the phosphate backbone orientated outwards. The binding of the substrate along the cleft of the two lobes catalyses the transfer of the terminal γ-phosphate of ATP to the hydroxyl oxygen of the Ser, Thr or Tyr residue of the substrate.6In addition to the kinase domain it has been observed that most of the human kinases hold a wide range of other functional domains which play major roles in regulating the activity of a given kinase. In the classical work of Hanks and Hunter7 the entire protein kinase family has been classified into groups and these groups are further classified into various families and subfamilies. Classification of a protein kinase into a subfamily indicates its function, its potential mode of regulation and the signal transduction pathway in which it is likely to participate. This review focuses on the available information on the human kinase with particular reference to the current understanding of the domain combinations observed in the kinase subfamilies. We also highlight the importance of regulation of kinases by the domains tethered to catalytic kinase domain and the relationship with diseases, focusing on the various disease causing mutations observed in the domains associated with kinase domain.
By and large it is observed that a given kinase subfamily type is consistent with a specific domain architecture. However in recent genomic studies, two types of outliers have been proposed which do not adhere to this rule and are not assigned unambiguously to a specific subfamily.8–10 The outlier kinases (which do not have domain architectures consistent with the subfamily type) have been classified into two categories, the “Hybrid kinases” and the “Rogue kinases”. A “Hybrid” kinase is one with the kinase catalytic domain having characteristic features of a kinase subfamily whereas the non-kinase domains in the same protein are characteristic feature of another kinase subfamily. A rogue kinase is one where the non kinase domain(s) or their sequential order (architecture) is unique and usually not observed among currently known Ser/Thr/Tyr kinases.8 This review provides an analysis of such outlier kinases seen among the human kinases and their evolutionary importance is highlighted by analyzing their counterparts in closely related primate species.
Multi-domain nature of human kinases
Cellular information processing involving reversible protein phosphorylation requires tight control of the localization, activity, and substrate specificity of protein kinases, which to a large extent is accomplished by the various domains tethered to the kinases. The human kinome can be subdivided into seven major groups according to the sequence and structure of the catalytic domain, including TK, Tkl (tyrosine kinase-like), Ste (homologues of the yeast sterile 7, sterile 11 and sterile 20 kinases), CK1 (casein kinase 1), AGC (family of protein kinases A, G and C), CAMKs (calcium/calmodulin-dependent protein kinases) and the highly conserved CMGC subgroup, which contains GSK3, CK, cyclin-dependent kinases (CDKs) and mitogen-activated protein kinases (MAPKs).11 These groups are further classified into various families and subfamilies based on the domain structure outside the catalytic domain.AGC group of kinases
AGC kinases are involved in diverse cellular functions and are potential targets for the treatment of human diseases such as cancer, diabetes, obesity, neurological disorders, inflammation and viral infections.12 These kinases have been classified into 22 subfamilies (Fig. 1).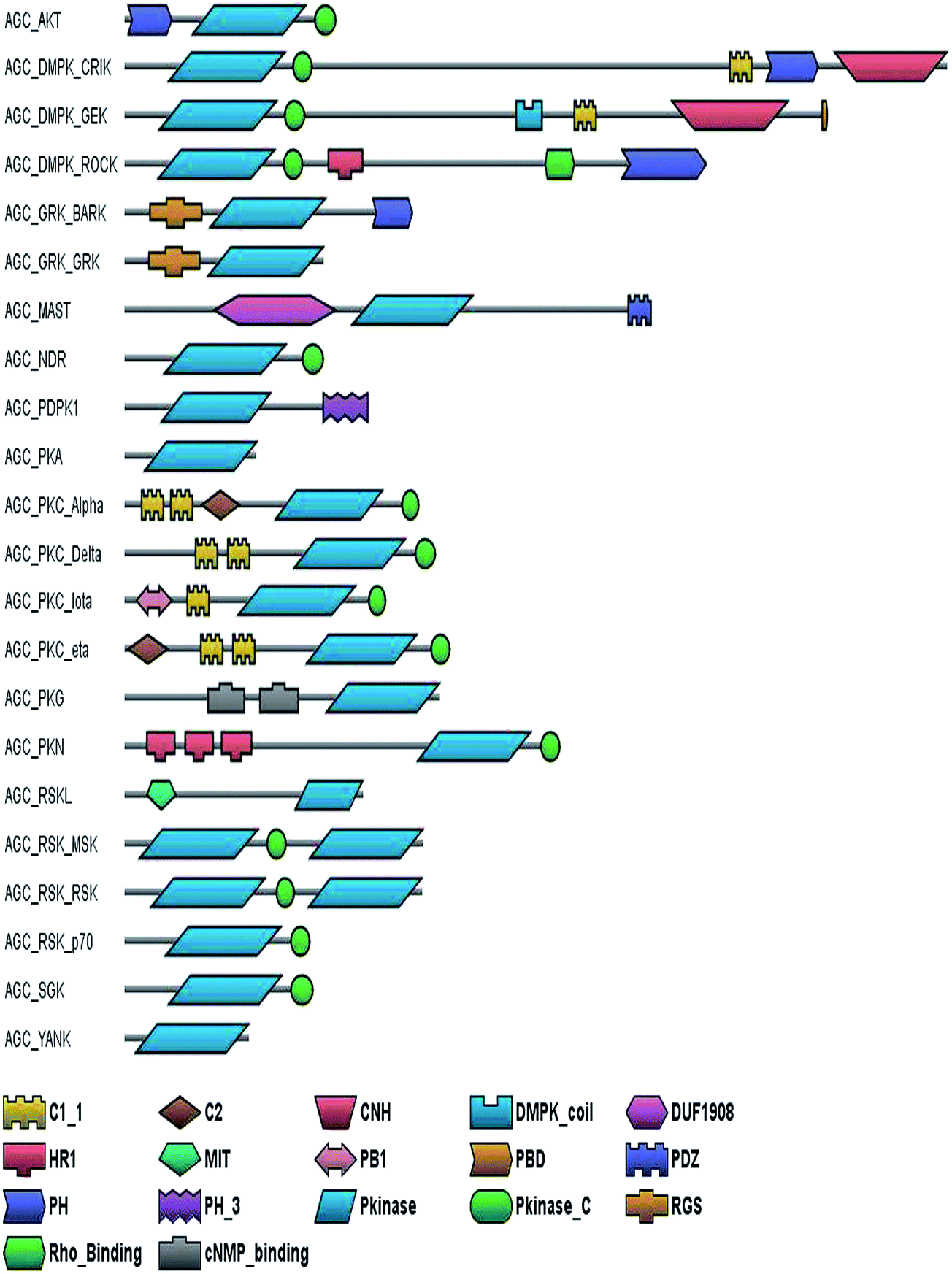 | ||
| Fig. 1 Canonical domain architecture of AGC kinases: the domain architecture of the different subfamilies of the AGC group of kinases have been shown considering one representative sequence from each subfamily. C1_1, conserved region; C2, conserved region CNH, citron homology; DMPK_coil, myotonin protein kinase; DUF1908, domain of unknown function 1908; HR1, heptapetide repeat 1; MIT, microtubule-interacting and trafficking; PB1, a protein interaction module; PBD, p21 Rho-binding domain; PDZ, common structural domain; PH, pleckstrin homology; Pkinase, protein kinase; Pkinase_C, protein kinase C-terminal; Rho_Binding, Rho-binding domain; RGS, regulator of G protein signalling; cNMP_Binding; cNMP binding domain. | ||
Akt isoforms possess an N-terminal pleckstrin homology (PH) domain which binds to phosphatidylinositol-3,4,5-trisphosphate (PtdIns(3,4,5)P3) and phosphatidyl inositol-3,4-bisphosphate (PtdIns(3,4)P2). This interaction of Akt isoforms leads to the activation of these kinases by PDK1.13 SGK kinases possess a phosphoinositide-binding Phox homology (PX) domain at its N terminus interacts with phosphatidylinositol-3-phosphate (PtdIns(3)P) and mediates the endosomal association of these kinases, that is essential for its phosphorylation and activation by PDK1.14 The classical PKC kinases interact with diacylglycerol and calcium by its N-terminal conserved region 1 (C1) and C2 domains, which results in a conformational change in these proteins that in turn mediate their activation.15
PKNs possess three leucine zipper-like heptapeptide repeat domains at their N-terminus, which interact with Rho–GTP and control their phosphorylation by PDK1.16 RSK and MSK are unusual kinases with two kinase catalytic domains; the N-terminal kinase domain (NTKD) being an AGC kinase and a C-terminal kinase domain (CTKD) which belongs to the calcium/calmodulin dependent protein kinase family.17 PDK1 kinase which is thought to phosphorylate more than 23 AGC kinases, localizes at the plasma membrane through its C-terminal PH domain.13 GRK kinases modulate the activity of the GPCR proteins and the binding of these kinases to the GPCRs is mediated by the RGS domain.18 Rho–GTP binds to the Rho-binding domain (RBD) of the ROCK and CRIK kinases and DMPK interacts with Rac–GTP.19 The ROCK, CRIK, MRCK and DMPK subfamilies have PH domains, which probably stabilizes their association with the membrane. Microtubule-associated Ser/Thr kinases (MAST) contain a PDZ domain and a domain of unknown function 1908 (DUF1908), in addition to the kinase domain. However the functions of these domains have not been studied extensively.20
CAMK group
In the CAMK group, members of the MLCK, RAD53, PKD, CAMK2, Trio, DCAMKL, CASK, DAPK and CAMKL subfamilies are multi-domain in nature (Fig. 2). Beyond the common kinase domain, the DAPK kinase, contains a long C-terminal extension with multiple functional domains. A series of eight ankyrin repeats follows the catalytic domain which is followed by a region that has been shown to direct the kinase to the actin cytoskeleton. The C terminus of DAPK contains a death domain, followed by a 17 residue tail rich in Ser residues, a feature common to other death domain-containing proteins which are critical regulatory segments of the DAPK kinases.21 The DCAMKL kinases contain two double cortin (DCX) domains followed by a kinase domain which plays a role in neuronal migration.22 CASK (Ca2+/calmodulin dependent serine kinase) which plays a key role in establishing inter-cellular contacts and plasticity at cellular junctions23 contains a series of L27 repeats, PDZ, two Src homology 3 (SH3) domains and a C-terminal guanylate kinase domain. L27 domain and the PDZ domain serve as protein interaction modules to co-ordinate interactions of CASK as a scaffolding complex and serving as a link between extracellular matrix and the actin cytoskeleton.24 The Trio kinase in par with its name holds three enzymatic domains, two functional GEF domains and a kinase domain. One of the GEF domains has a Rac specific activity and the other domain has a Rho specific activity. In addition the C-terminal contains Ig domain and N-terminal has four spectrin like repeats that play a key role is intracellular targeting.25 The CAMKII holds a self-association domain (CaMKII AD) found at the C terminus, which helps in the assembly of the single proteins into large (8 to 14 subunits) multimers.26 PKD has a putative PH domain that is reminiscent of those found in the protein kinase B family and is important for regulating PKD's enzyme activity in addition to two C1 domains which bind to DAG and phorbol esters.27 In the case of Rad53 kinase, it has been demonstrated that association of the C-terminal FHA2 domain with phosphorylated Rad9 is essential for Rad53 activation and G2/M arrest following DNA damage.28 In addition to its kinase activity, the myosin light chain kinase (MLCK) of smooth muscle has an actin binding activity through which it can regulate the actin–myosin interaction of smooth muscle.29 The titin kinase whose kinase domain has a high similarity with the MLCK kinase domain is sandwiched by several immunoglobulin fibronectin type III-like domains.30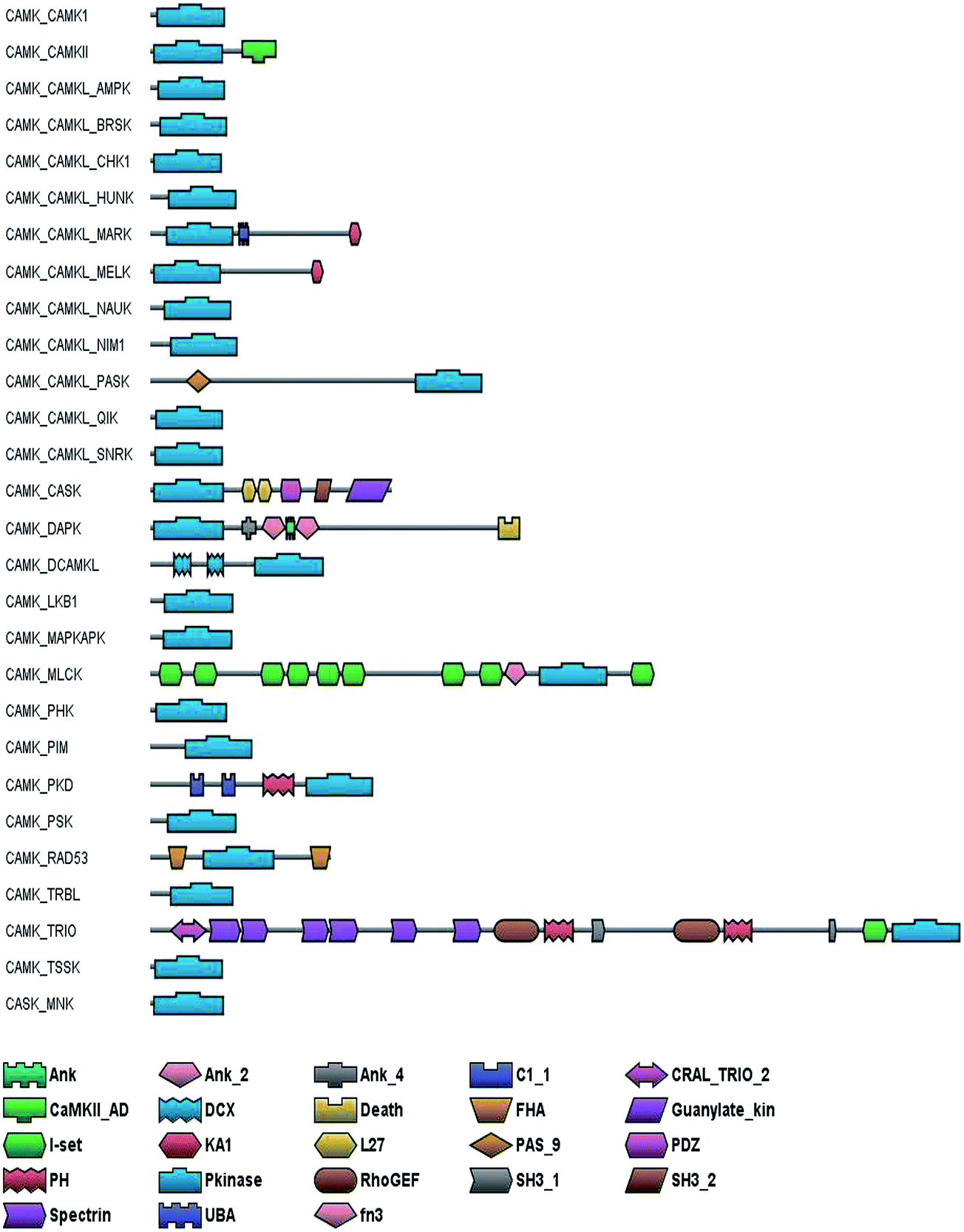 | ||
| Fig. 2 Canonical domain architecture of CAMK kinases: the domain architecture of the different subfamilies of the CAMK group of kinases have been shown considering one representative sequence from each subfamily. Ank, ankyrin repeat; Ank_2, ankyrin repeat 2; Ank_4, ankyrin repeat 4; C1_1, conserved region; CRAL_Trio_2, named after cellular retinaldehyde-binding protein (CRALBP) and TRIO guanine exchange factor; CAMKII_AD, CamKII adhesion domain; DCX, doublecortin; Death, death associated domain; FHA, forkhead-associated domain; Guanylate_kin, guanylate kinase; I set, immunoglobulin domain; KA1, kinase associated domain 1; L27, protein interaction module; PAS_9, pas domain; PDZ, common structural domain; PH, pleckstrin homology; Pkinase, protein kinase; RhoGEF, structural domain of guanine nucleotide exchange factors for Rho/Rac/Cdc42-like GTPases; SH3_1, SH3_2, SRC homology 3 domain; Spectrin, spectrin repeat; UBA, ubiquitin binding domain; fn3, fibronectin type III domain. | ||
CK1 group
The CK1 group forms a unique and small branch of the protein kinase family, populated majorly with the CK1 gene products along with their closest relative tau–tubulin binding kinases and the vaccinia-related kinases.31 Kinases in this group do not hold additional non-catalytic domains outside the kinase domain except for the CK1 gamma subfamily which holds a CK1 gamma domain whose function is yet unknown.CMGC group
CMGC group of kinases comprises of dual-specificity tyrosine-regulated kinases, or dual-specificity yak-related kinases (DYRKs), cyclin-dependent kinases (CDKs), mitogen-activated protein kinases (MAPKs), glycogen synthase kinase-3 (GSK3), CDK-like kinases, serine/arginine-rich protein kinases, cdc2-like kinases, and RCK kinases.32 Similar to the CK1 group the members of the CMGC group members are single domain kinases.TK group
The mammalian non-receptor tyrosine kinases (NRTKs) are divided into ten families: Src, Abl, Jak, Ack, Csk, Fak, Fes, Frk/Fyn, Tec, and Syk.33 In addition to their tyrosine kinase catalytic domains, they all contain non-catalytic domains that are important in enzyme regulation and substrate recognition34 (Fig. 3A). Src family kinases have a conserved domain architecture consisting of unique regions which are followed by SH3, SH2, and kinase catalytic domains. The SH2 and SH3 domains are critical for targeting the Src kinases to their appropriate cellular locations and also the regulation of the Src kinases.35 The SH3–SH2–kinase domain arrangement is conserved in other families of non-receptor tyrosine kinases such as Csk and Fer subfamilies playing similar roles. The Abl family of kinases have a domain organization similar to that of the Src family kinases, with sequential Src homology SH3 and SH2 domains preceding the kinase domain. In addition to SH2 and SH3 domains, Abl possesses an F-actin-binding domain and a DNA-binding region.36 Fak possesses two domains that mediate protein–protein interactions: a ferm domain and a focal adhesion-binding domain.37 Another modular domain, present in the Btk/Tec subfamily of NRTKs and in many other signaling proteins, is the PH domain. PH domains mediate the binding of these kinases to activated single complexes at the membrane by interacting with phosphorylated phosphatidylinositol lipids.38 The Fes family of proteins hold a Fes/CIP4, and EFC/F-BAR homology domain followed by SH2 domain. Coordinated actions of the F-BAR and SH2 domains have been found to regulate the actin reorganization in mast cells by the coupling to FcεRI signaling.39 Tandem SH2 domains are found in members of the SYK subfamily. Engagement of these SH2 domains by tyrosine-phosphorylated immunoreceptor tyrosine-based activation motifs leads to kinase activation and downstream signaling.40 | ||
| Fig. 3 Canonical domain architecture of tyrosine kinases: the domain architecture of the different subfamilies of the tyrosine group of kinases have been shown considering one representative sequence from each subfamily. (A) Non-receptor tyrosine kinases. BTK, Btk-type zinc finger; FCH, Fes/CIP4, and EFC/F-BAR homology domain; FERM_M, ferm domain; F-actin_bind, F-actin binding domain; Focal_AT, focal adhesion targeting protein; GTPase_binding, GTPase binding domain; Inhibitor_Mig-6, EGFR receptor inhibitor Mig6; PH, pleckstrin homology; Pkinase_Tyr, protein tyrosine kinase; SH2, SRC homology 2 domain; SH3_1, SH3_9, SRC homology 3 domain; UBA, ubiquitin binding domain; (B) receptor tyrosine kinases. EphA2_TM, ephrin type-A receptor 2 transmembrane domain; Ephrin_lbd, ephrin receptor ligand binding domain; F5_F8_type_C, discoidin domain; Furin-like, furin-like cysteine rich region; Fz, Fz domain; GCC2_GCC3; GF_recep_IV, growth factor receptor domain IV; Gly_rich, glycine rich; I-set, Ig2, IgTie2_1, immunoglobulin domain; Kringle, kringle domain; LRR8, leucine rich repeat; Ldl_recept_a, low-density lipoprotein receptor domain class A; MAM, MAM domain; PSI, photosystem I reaction centre; Pkinase_Tyr, protein tyrosine kinase; SAM_1, sterile alpha motif; Sema, semaphorin structural domain; TIG, IPT/TIG domain; TM, transmembrane region; V-set, immunoglobulin V-set domain; WIF, WIF domain; fn3, fibronectin type III domain; hEGF, human growth factor-like EGF. | ||
Receptor tyrosine kinases (RTKs) play essential roles in cellular processes, including metabolism, cell-cycle control, survival, proliferation, motility and differentiation.41 They have been classified into 20 different subfamilies (Fig. 3B). The Ig-like domain is the most common domain type found in the RTK ectodomains occurring in the platelet-derived growth factor receptor PDGFR subfamily, vascular epithelial growth factor receptor VEGFR subfamily, protein tyrosine kinase 7 PTK7, nerve growth factor receptor Trk subfamily, receptor tyrosine kinase-like orphan receptor Ror subfamily, muscle-specific kinase Musk subfamily, Met subfamily, Axl subfamily and Tie subfamily.42 These domains act as ligand binding sites for a number of growth receptors.43 Members of the InsR family each contain three FnIII (fibronectin type III domain), which is also seen in the members of the Axl family Tie family and the Ros subfamily.33 A single Eph domain occurs in each member of the ephrin-interacting type A and type B receptors (namely, EphA1-8, EphA10, EphB1-4 and EphB6).44 Discoidin domains (F8_F5_type_C) occur in the DDR subfamily of RTKs that binds to collagen-II.45 The Mam domain in one RTK family, namely, the anaplastic lymphoma kinase (ALK) family regulates the kinase activity of the members in this subfamily.46 The Ror family of RTKs have two kinds of domains namely frizzled and kringle domains in addition to Ig repeats, both of which are assumed to mediate protein–protein interactions.47 The RYK subfamily has a WIF domain (Wnt-inhibitory factor) that helps in dimerisation with transmembrane proteins.48 The ligand binding domain of the ephrin receptors tyrosine kinases (EphB) which comprise of the ephrin binding domain are also followed by cysteine-rich regions referred to as GCC2–GCC3 repeats and the fn3 repeats.49
TKL group
The TKL group comprises of 8 major subfamilies (Fig. 4). The IRAK subfamily members have a death domain at their C-terminal end. The death domain (DD) of IRAK-1 mediates the interaction with other molecules of the signaling complex, e.g., the adaptor MyD88, the silencer Tollip, and the activator kinase IRAK-4.50 The LIM (Lin11/Is11/Mec3) domain is found associated with the LIMK kinase subfamily which mediates protein–protein interactions.51 LRRK subfamily members are large proteins encoding multiple functional domains, including two catalytically active domains, a kinase and a GTPase domain in addition to multiple LRR repeats.52 The MLK kinases have a C-terminal SH3 domain which binds to dynamin and enhances the activation of GTPase.53 The Raf family of proteins contain a Ras-binding domain (RBD), which is necessary for the interaction with Ras and with membrane phospholipids required for membrane recruitment54 and an important inhibitory phosphorylation sites participating in the negative regulation of Ras binding and Raf activation.55 The STKR subfamily of proteins bind to their ligands activin and TGFβ with the help of the respective domains seen in the N-terminal end of the kinase domain.56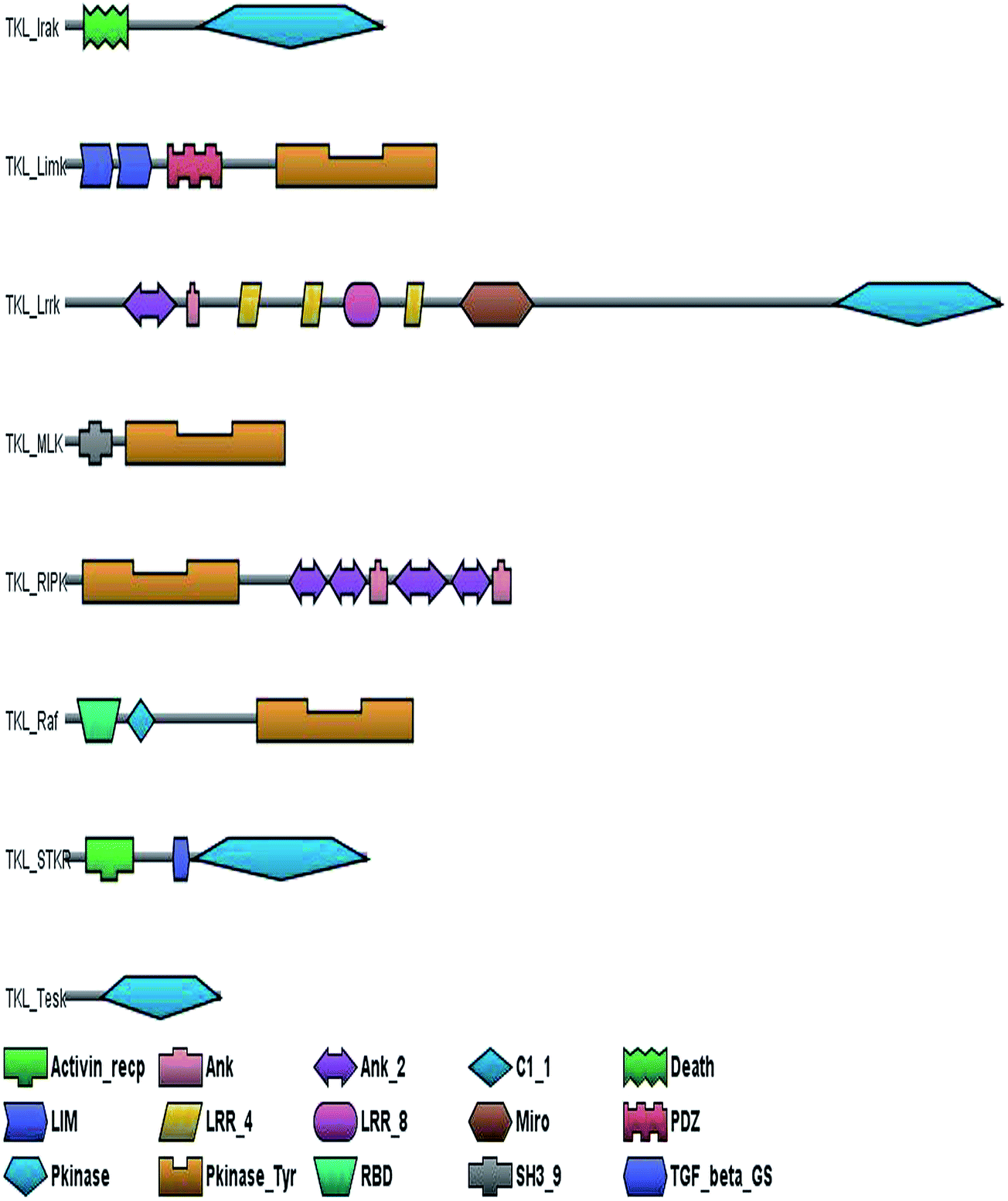 | ||
| Fig. 4 Canonical domain architecture of receptor tyrosine kinase like (TKL) kinases: the domain architecture of TKLs have been shown considering one representative sequence from each subfamily. Activin_recep, activin receptor domain; Ank, Ank_2, ankyrin repeat; C1_1, conserved region; Death, death associated domain; LIM, lim domain; LRR_4, LRR_8 leucine rich repeat; Miro, mitochondrial Rho proteins; PDZ, common structural domain; Pkinase, protein kinase; Pkinase_Tyr, protein tyrosine kinase; RBD, Rho binding domain; SH3_9, SRC homology 3 domain; TGF_beta_GS, transforming growth factor beta (TGF-beta) type I GSmotif. | ||
STE group
Most of the kinases in the STE group comprise only of kinase domain while some of them have additional regulatory domains (Fig. 5) The Ask subfamily of STE11 kinases and the MSN subfamily of the STE20 have a CNH binding domain which interacts with the small GTPase and regulates the actin cytoskeleton. The PAK subfamily of the STE20 kinases have a PBD domain that binds to cdc42–GTPases activating the signaling cascade which act upstream in the MAPK cascade.57 The FRAY subfamily of kinases consist of a OSR1 domain which is thought to bind to WNK1 and NKCC1.58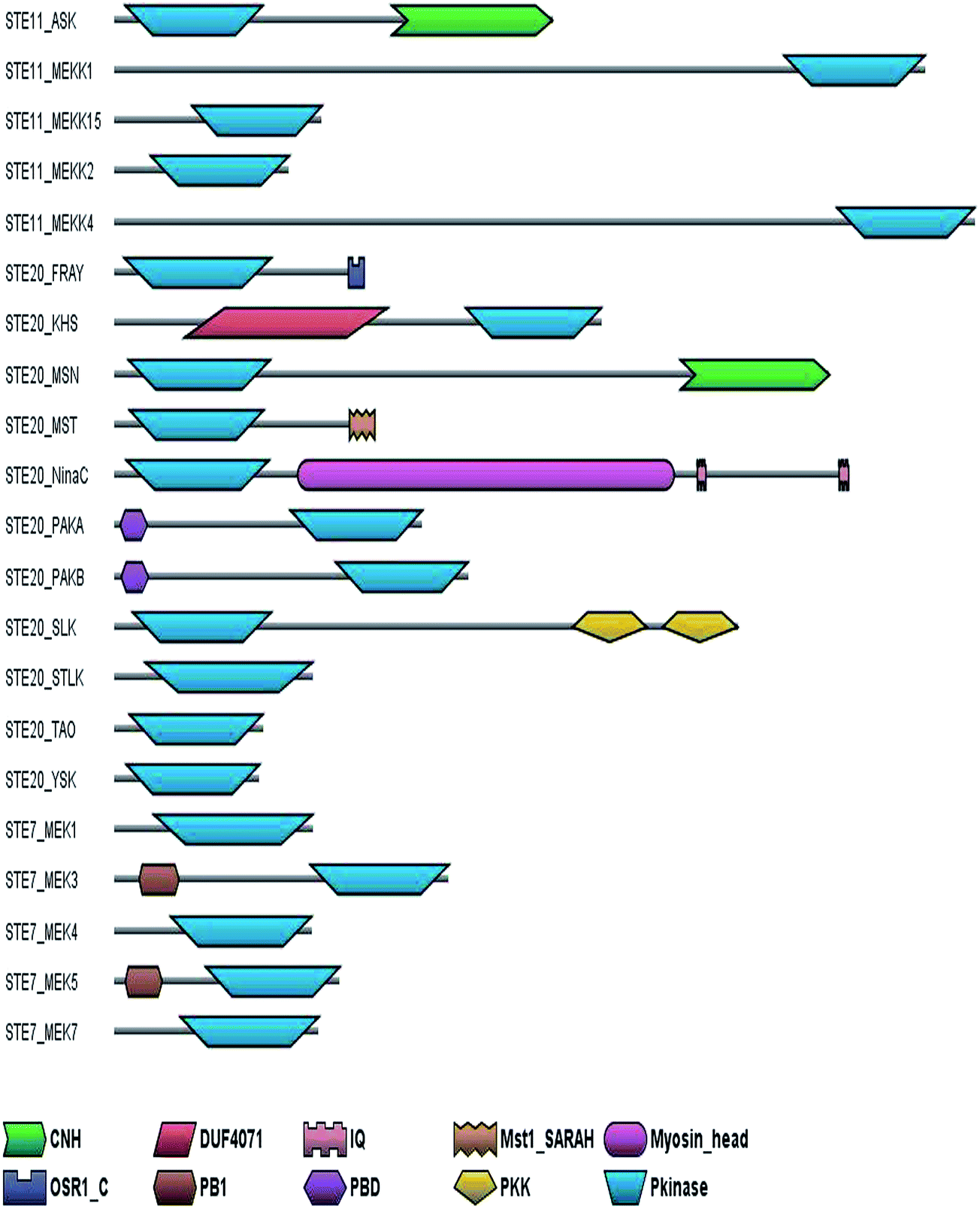 | ||
| Fig. 5 Canonical domain architecture of receptor STE group of kinases: the domain architecture of the different subfamilies of the STE group have been shown considering one representative sequence from each subfamily. CNH, citron homology; DUF4071, domain of unknown function 4071; IQ, IQ calmodulin-binding motif; Mst1_SARAH, C terminal SARAH domain of Mst1; Myosin_head, myosin head (motor domain); OSR1_C, oxidative stress responsive kinase 1 C-terminal; PB1, a protein interaction module; PBD, p21 Rho-binding domain; PKK, polo kinase kinase; Pkinase, protein kinase. | ||
Mutations outside kinase domains leading to disease states
Human kinases are known to phosphorylate about ∼30% of all human proteins involved in major cellular processes, such as growth, differentiation, proliferation and apoptosis.3 Given the importance of these cellular activities, the catalytic activity of kinases involved in these pathways is stringently regulated. Mutations in kinase genes have been identified to play major roles in a number of human diseases such as developmental and metabolic disorders, as well as certain cancers.4 Although a major part of such mutations are found to be localized in the kinase catalytic domain, some of them have been identified in other regulatory domains in case of the multi-domain kinases some of which are described below.Akt kinase, also known as protein kinase B, plays key roles in cell proliferation, survival and metabolism. The Akt isoforms binds PIP3 through its PH domain, resulting in translocation of Akt to the membrane. A somatic mutation that results in the E17K substitution in the PH domain of the AKT1 has been identified in human breast, colorectal, and ovarian cancers59 and Proteus syndrome.60 Lys17 alters the electrostatic interactions of the pocket and forms new hydrogen bonds with a phosphoinositide ligand. This mutation activates AKT1 by means of pathologic localization to the plasma membrane, stimulates downstream signaling, transforms cells, and induces leukemia.59 In 3 patients with hypoinsulinemic hypoglycemia and hemihypertrophy, Hussain et al. identified a similar glu17-to-lys (E17K) substitution in the PH domain of the AKT2 gene.61 The same mutation in AKT3 gene was found in patients with hemimegalencephaly (HME) which is an overgrowth syndrome.62,63
Six mutations that occur in the conserved C1 regulatory domain of the PRKCG have been identified in patients with spinocerebellar ataxia.64–68 Localized solely in the brain and spinal cord, the PRKCG has been demonstrated to be important for several neuronal functions, including long term potentiation (LTP) and long term depression (LTD). Mutations in the C1 regulatory domain were found to severely abrogate the zinc-binding or phorbol ester-binding capabilities of the protein.
PRKG1 gene plays important roles in physiologic processes such as relaxation of vascular smooth muscle and inhibition of platelet aggregation. Patients with autosomal dominant thoracic aortic aneurysm were identified with an R177Q substitution at a highly conserved residue in the CNB-A domain of the PRKG1 gene.69,70 Activation of the PRKAR1A gene occurs when 2 cAMP molecules bind to each regulatory subunit (cAMP binding domains), eliciting a reversible conformational change that releases active catalytic subunits.71 identified mutations in cAMP-binding domain B of the PRKAR1A gene in patients with Acrodysostosis 1 and suggested that the mutation would cause reduced cAMP binding, reduced PKA activation, and hence decreased downstream signaling.
Titin, or connectin, the giant muscle protein expressed in the cardiac and skeletal muscles spans half of the sarcomere and plays a key role in muscle assembly, force transmission at the Z line, and maintenance of resting tension in the I band region.72 The A743V mutation located in the alpha-actinin binding domain of titin, decreases the affinity of titin Z-repeats to alpha-actinin by about 40% compared to normal resulting in cardiomyopathy. Another mutation of V54M located in the telethonin binding domain of titin, showed that the mutation decreased the affinity of titin for telethonin to about 60% of normal. Q4053X and S4465N substitution of the N2-B domain, which is known to be expressed only in cardiac muscle was also associated with cardiomyopathy.72
Centronuclear myopathy 5 showed a substitution of Gly2757Val in the second fibronectin type III domain of tSPEG.73 The MLCK protein contains 9 C2-type immunoglobulin-like homology domains, an SH2-binding domain, and a single tyrosine phosphorylation site in the CaM-binding region. In 3 affected members of a family with aortic aneurysm and dissection Wang et al. identified heterozygosity for a 5275T-C transition in the MYLK gene, resulting in a Ser1759 to Pro substitution in the alpha-helix of the calmodulin-binding sequence that was predicted to cause loss of MLCK function by altering calmodulin binding.74 CHK2, a protein kinase that is activated in response to DNA damage, is involved in cell cycle arrest. An I157T mutation within the forkhead homology-association domain of CHK2 resulting in Li–Fraumeni syndrome variant associated with the development of three primary tumors: breast cancer, melanoma, and lung cancer.75 FG syndrome 4 is characterized by R28L substitution at a highly conserved residue in the CaM-kinase domain of the CASK gene.76
R206H77–80 and R202I81 substitution at the end of glycine–serine (GS) activation domain of the ACVR1 gene was identified in patients with fibrodysplasia ossificans progressiva. Parkinson disease 8 is characterized by a R1441G/H/C substitutions in the RAS domain of LRRK2 gene and R1441C81–84 R1441H. S257L and P261S substitution at the CR2 domain of RAF1 gene was identified in Noonan syndrome 5 (ref. 82 and 83) where both the mutations showed an increased kinase and ERK activation. T491R substitution in the CR3 domain of the RAF1 gene was also observed with patients with Noonan syndrome 5.82 L603P substitution at this domain was identified in patients with cardiomyopathy.84
Chronic myelogenous leukemia (CML) is a hematopoietic stem cell disorder characterized by the Philadelphia chromosome, the result of a (9;22) translocation that fuses BCR sequence with the ABL gene and produces the constitutively active, Bcr–Abl tyrosine kinase. Point mutations in the kinase domain of BCR–ABL are the most common mechanism of drug resistance in chronic myeloid leukemia (CML) patients treated with ABL kinase inhibitors, including imatinib. It has also been shown in vitro that mutations outside the kinase domain in the neighboring linker, SH2, SH3, and Cap domains can confer imatinib resistance. T315I substitution is considered a major cause of resistance to imatinib as Thr315 is important to form a hydrogen bond with the secondary amino group of imatinib.85
In a patient with atypical X-linked agammaglobulinemia,86 found a point mutation Y361C in the SH2 domain of BTK in a B-cell line which is a key regulator of B-cell development. Agammaglobulinemia is an immunodeficiency disorder characterized by failure to produce mature B lymphocytes and associated with a failure of Ig heavy chain rearrangement.87 SH2 domains are critical mediators of binding with phosphotyrosine-containing proteins in the cell. P33T and D113V mutations located in the PH domain of BTK was also observed in some patients.88 Some patients with the same disease phenotype showed mutation in the SH3 domain.88,89 Spondylometaepiphyseal dysplasia patients showed E113K mutation located in the extracellular discoidin domain which affects a surface-exposed residue important in the DDR2 collagen-binding site.90
G948W and T940I substitution in the cytoplasmic sterile-alpha-motif (SAM) domain of the EPHA2 gene was found in patients with cataract.91,92 The FGFR tyrosine kinase members bind to the fibroblast growth factors, and mutations in FGFR1, FGFR2, and FGFR3 that are associated with distinct clinical entities, including achondroplasia, hypochondroplasia, thanatophoric dysplasia, Antley–Bixler syndrome, Apert syndrome, Beare–Stevenson syndrome, Crouzon syndrome, Jackson–Weiss syndrome, Pfeiffer syndrome, and Saethre–Chotzen syndrome.93 G237S substitution in the Ig-like domain D2,94 G48S mutation in the IgI domain95 and G348R substitution in the D3 domain96 of the FGFR1 gene were identified in patients with idiopathic hypogonadotropic hypogonadism. C342Y substitution within the third Ig domain of FGFR2 gene has been identified as the leading causes of Crouzon syndrome.97,98 Apart from this Y328C and S347C in the same domain have been identified in patients with Crouzon syndrome. Achondroplasia is characterized by the S279C mutation in FGFR3 at the IgIIIa extracellular domain. Y278C and S84L mutations have been observed in the extracellular domains of FGFR3 in the patients with hypochondroplasia.99 A855T substitution in the ATP-binding domain of FLT4 receptor was seen in patients with lymphedema.100 Lymphoproliferative syndrome is characterized by mutations R335W in the BG loop of the SH2 domain101 and R29H substitution in the PH domain of ITK.102
Unusual domain architectures
As mentioned earlier the major kinase subgroups described by Hanks and Hunter, based on the kinase catalytic domain, are further classified into various kinase subfamilies. These kinase subfamilies show several domain combinations each of which is related to the regulation and function of kinases belonging to that particular subfamily. However interestingly in a few rare instances, it is seen that there is mix and match of these domain combinations, wherein a particular kinase annotated to belong to specific subfamily of kinase on the basis of its kinase catalytic domain, has a domain architecture characteristic of another subfamily of kinase. Such kinases may be broadly classified into two categories; hybrid and rogue kinases.8Description of a hybrid kinase
Let us consider two classical sub-families of kinases A and B with the characteristic property of the kinase subfamily A is to tether with a domain family P while the kinase subfamily B typically tethers with domain family Q. However if we find a kinase with sequence of kinase catalytic domain clearly characteristic of kinase subfamily A, but it tethers with a domain which belongs to domain family Q then such a sequence has chimeric or hybrid properties of two different subfamilies of kinases i.e., it has the features of kinase subfamily A in the catalytic domain while the associated non-kinase domain is a characteristic feature of kinase subfamily B (Fig. 6A). Therefore it is inappropriate to classify such a kinase into kinase subfamily A or B and it should be more appropriately referred as a “hybrid kinase” with features of two different kinase subfamilies shared. Therefore one would expect some of the properties such as substrate specificity or substrate recruitment or mode of regulation or sub-cellular localization are different from that of the classical kinase subfamily. Indeed such hybrid kinases may facilitate cross talks between signaling pathways. | ||
| Fig. 6 Hybrid and rogue kinases: mix and match of catalytic kinase domain and tethered domain architectures to yield outlier kinases (A) hybrid and (B) rogue. | ||
Description of a rogue kinase
Let us consider a classical kinase subfamily A with a characteristic property of this subfamily to tether with a domain family P. However if we find a kinase with sequence of catalytic domain characteristic of kinase subfamily A, but it is tethered to domain family R (Fig. 6B) which is not typical of any known kinase subfamily then this represents a rogue kinase with functional or regulatory characteristics different from that of the classical subfamily A.Hybrid kinases
Hybrid kinases correspond to the cases where the kinase catalytic domain shows good similarity to a specific subfamily of kinases while the domain architecture is characteristic of another kinase subfamily. And in other instances they are single domain kinases but their classified subfamilies are typical of multi-domain kinases with specific domain architectures. There are 14 sequences with domain combinations that are unusual in the context of their particular kinase subfamilies identified solely on the basis of kinase catalytic domain. These are listed in Table 1 with details of the domain combination normally seen for the subfamily assignment, the observed domain combination in the hybrid sequences and their corresponding subfamily. The importance of such domain combinations are further discussed below.| Protein ID | Group | Subfamily | Normal domain association | Observed domain association | Cognate kinase subfamily corresponding to the observed domain |
|---|---|---|---|---|---|
| Q9NRM7 | AGC | NDR | Single kinase domain | UBA | CAMKL (CAMK) |
| O95835 | AGC | NDR | Single kinase domain | UBA | CAMKL (CAMK) |
| P34947 | AGC | GRK | RGC | PH | AKT, CRIC, ROCK (AGC) PKD (CAMK) |
| P26098 | AGC | GRK | RGC | PH | AKT, CRIC, ROCK (AGC) PKD (CAMK) |
| Q9C098 | CAMK | DCAMKL | DCX | Single kinase domain | |
| Q9UIK4 | CAMK | DAPK | Ank, death domain | Single kinase domain | |
| Q32MK0 | CAMK | MLCK | I-set, Fn3 | Single kinase domain | |
| Q9H1R3 | CAMK | MLCK | I-set, Fn3 | Single kinase domain | |
| A86YV6 | CAMK | MLCK | I-set, Fn3 | Single kinase domain | |
| Q6VAB6 | TKL | RAF | RBD, C1 | SAM | Eph (TK) |
| Q8IVT5 | TKL | RAF | RBD, C1 | SAM | Eph (TK) |
| P29376 | TK | ALK | Ldl_receptor, MAM | No Ldl_receptor, MAM but Gly_rich domain | Atypical |
| P42681 | TK | TEC | PH, BTK, SH3, SH2 | Only SH3, SH2 | Csk, Fyn, Src (TK) |
| Q13470 | TK | Ack | SH3, GTPase, MIg6, UBA | Only SH3 domain | Abl, Src, Csk (TK) |
G protein-coupled receptor kinases (GRKs) constitute a family of seven serine/threonine protein kinases that specifically recognize and phosphorylate agonist-activated G protein-coupled receptors (GPCRs). The GRK kinases have been shown to have an RGS domain (regulator of G protein signalling homology domain) which plays an important role in regulation of these kinases. However exceptionally it is seen that GRK2 and GRK3 contains a PH domain with binding sites for the membrane phospholipid PIP2 and free Gβγ subunits.103 Since GRK2 and 3 are cytosolic proteins, these specific interactions could help to maintain a membrane-bound population of GRK2 prior to the agonist-dependent GRK2 translocation.104
The human genome encodes four related kinases, NDR1, NDR2, LATS1 (large tumour suppressor-1) and LATS2 belonging to the NDR subfamily. This subfamily is characterized by a single kinase domain with an exception of LATS1 and LATS2 which are tethered to a UBA domain on their N-terminal region. The presence of UBA domain was thought be restricted to the AMPK related kinases where they play an important role in regulating the conformation, activation and localization of these enzymes.105 Similar roles of the UBA domain in the LATS1 has been reported recently where the deletion of the N-terminal segment of this protein was found to suppress tumor suggesting that they could also play an important role is the regulation of the LATS kinases.106
Doublecortin-like kinases (DCLK) are widely expressed in postmitotic neurons throughout the embryonic nervous system. The N-terminal doublecortin domain helps in the localization of the kinases to microtubules.107 However the DCLK3 does not include this domain.
The KSR1 and KSR2 members of the TLK RAF subfamily comprise of SAM domain while the other members comprise of RBD binding domain. It has been shown that the SAM domain helps in the localization of these kinases to specific sites in the plasma membrane.108
The members of the MLCK subfamily in general are characterized by the presence of Ig repeats and fibronectin type 3 like repeats which serve as molecular spring and help in the regulation of dynamics of the filaments that constitute contractile proteins.109 Such features are not present in MLCK 2, 3 and 4 which are single kinase domain proteins.
Similarly the TNK1 kinase that belongs to the Ack subfamily does not have the canonical domain architecture of its other subfamily member which comprise of SH3, CRIB. Mig6 and UBA domain but retains only the SH3 domain.
Rogue kinases
The rogue kinases are those cases which show domain architectures not characteristic of any protein kinase subfamily though the catalytic domain sequence is characteristic of a protein kinase subfamily. The domain combinations which are considered as rogue kinases are shown in Fig. 7. The possible roles of these novel domain combinations have been discussed below in the light of domain architecture diagrams in Fig. 7. | ||
| Fig. 7 Rogue kinases with their homologues: human kinases with kinase catalytic domain belonging to a specific kinase subfamily, but, the tethered domain architecture is not characteristic of that subfamily or any other subfamily of kinases. The conservation of domain architecture of rouge kinases across other primates is represented in this figure. ATG16, Autophagy protein 16 (ATG16); CARD, caspase recruitment domain; DEATH, death associated domain; Mei4, meiosis-specific protein PX, phosphoinositide-binding structural domain; Pkinase, protein kinase. Pkinase_Tyr, protein tyrosine kinase; Pkinase_C, protein kinase C-terminal; RHIM, RIP homeotypic interaction motif; cNMP_Binding; cNMP binding domain; ecTbetaR2, transforming growth factor, beta receptor II. | ||
The Autophagy protein 16 (ATG16) domain110,111 is shown to be involved in autophagy.111 The catalytic domain of one of the rogue kinases shows similarity to cyclic-GMP dependent kinase. A previous study shows the loss of this kinase activity during immortalization.111 This indicates the importance of c-GMP regulated role of this kinase in cell survival.
The Zf-RING-like domain is involved in cytoskeleton remodelling.112 PKD kinases usually mediate extra-cellular receptor mediated signal transduction.113 This particular kinase has a PH domain suggesting its localization to the membrane. Therefore, this protein kinase may suggest a regulatory role in cytoskeleton remodelling.
The DAXX domain is implicated as a transcriptional repressor and is associated with DNA binding proteins.114 The kinase belongs to TTBK subfamily which is involved in amyloidogenesis. However the rogue kinase characterized by tethering of kinase domain with DAXX domain may be involved in a regulatory nature to control transcription of certain genes.
The Mei domain is associated with a family of meiosis specific proteins which are required for correct meiotic chromosome segregation and recombination.115 The association of this domain with the DAPK kinase which plays a role in cell death associated functions is interesting to note.
While all the other members of the STKR family of proteins in the TKL group comprise of an activin receptor domain and TGF (Transforming growth factor beta type I GS-motif) domains, TGFR2 includes the TGF beta receptor 2 domain which influences its ligand binding specificity.
The RIPK family of proteins do not show a conserved domain architecture as seen in the other subfamilies of kinases. RIPK1 contains a C-terminal death domain (DD) that also harbours a RIPK homotypic interaction motif (RHIM). RIPK2 also contains the N-terminal kinase domain and a C-terminal caspase activation and recruitment domain (CARD). Although RIP3 contains the N-terminal kinase domain, it has a unique C-terminal sequence that contains only a RHIM domain. In this analysis we have considered RIPK1 RIPK2 and RIPK3 as rouge kinases considering RIPK4 and RIPK5 which have the kinase domain and C-terminal ankyrin domains as the canonical architecture for this subfamily.
The unique domain combinations represent an elegant framework of variations in domain combinations to achieve variations in the overall function of the protein. This feature is especially interesting in the case of protein kinases since it shows varied domain combinations and regulates a large number of pathways.
Conservation across primate lineage
The presence of hybrid and rogue kinases in many eukaryotes has been established in the previous studies in this laboratory.8,9 This section specifically focuses on the homologues of human hybrid/rogue kinases in closely related primate species. Out of the 12 primate species whose genome sequence data is available, 5 datasets are well annotated which are appropriate for the current analysis.116 These are genome datasets of Chimpanzee, Gorilla, Orangutan, Gibbon and Bushbaby. The conserved domain architectures of the homologues are shown in Fig. 7 and 8. Most of the primate homologues have the hybrid/rogue domain architecture conserved. The sequence identity between the human hybrid/rogues and primate homologues is very high which implies very similar function of homologues as that of human hybrid/rogue kinases. Some of the hybrid kinases contribute to different ligand specificities, compared to canonical counterparts, or cross talks between signalling pathways by means of protein–protein interactions or recruiting UBA domains. The rogue kinases, by means of certain domain recruitments, lead to direct interaction with transcription machinery, changes in localization or cross talks between signalling pathways.8,9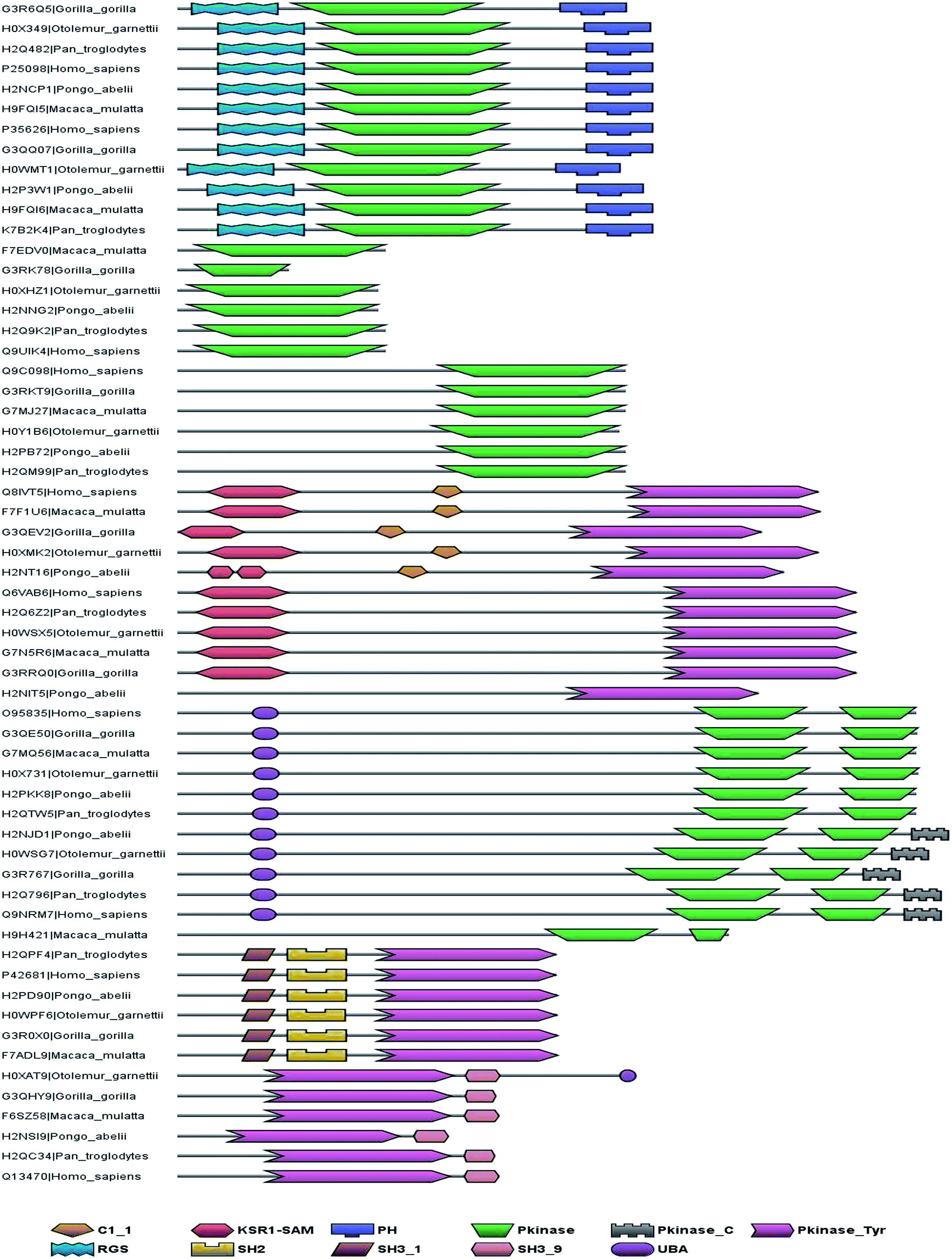 | ||
| Fig. 8 Hybrid kinases with their homologues: human kinases with kinase catalytic domain belonging to a specific kinase subfamily, but, the tethered domain architecture is not characteristic of that subfamily but to another subfamily of kinases. The conservation of domain architecture of hybrid kinases across other primates is represented in this figure. C1_1, conserved region; KSR1-SAM, SAM like domain present in kinase suppressor RAS 1; PH, pleckstrin homology; Pkinase, protein kinase; Pkinase_C, protein kinase C-terminal RGS, regulator of G protein signalling; SH2, SRC homology 2 domain; SH3_1, SH3_9, SRC homology 3 domain; UBA, ubiquitin binding domain. | ||
Discussion
This study reviews the effect of domain combination in dictating the overall function of a kinase specifically in the context of human kinases. Over the past 20 years, many mutations in kinase genes have been found to underlie several human diseases, particularly developmental and metabolic disorders, as well as certain cancers. Such mutations are sometimes located in the non-kinase domains of the protein and they have been discussed in this review further highlighting the importance of the additional domains in regulating the activity of a given kinase.The non-kinase domains and their sequential order are known to be related to the subfamily of the kinase domain. Indeed the classical approach proposed by Hanks and Hunter to classify kinases purely based on the sequence of kinase catalytic domain has resulted in a classification which is remarkably consistent with domain combinations in kinases.
The present study has shown the existence of outlier kinases at two levels with respect to particular subfamily (hybrid kinases) and also the protein kinase family as a whole (rogue kinases). The hybrid kinases reveal the possibility of mix and match of kinase and non-kinase domains potentially to elicit altered overall functions compared to canonical kinase subfamilies. The rogue kinases potentially help in rewiring certain proteins by simple domain recombination. Such kinases could facilitate cross-talk between two pathways as indicated in Fig. 9. Since a large amount of functional importance is encoded throughout the sequence and not just the kinase catalytic region, there is a need to classify kinases on the basis of full-length sequence which would supplement the classical Hanks and Hunter classification scheme. A clustering method and clues that would aid in such classification is provided in detail in earlier studies.8
 | ||
| Fig. 9 Cartoon representation of cross-talks between pathways mediated by rogue kinases. | ||
Human kinome study has been revisited by considering full-length of kinase sequences as most human kinases are multi-domain in nature. Since the full length sequences are to be studied in detail it was necessary that the gene definitions and sequences were up to date. Therefore, the latest version of human genome data was used in this study. Five primate species were also analyzed for the presence of hybrid and rogue kinases. Many species could be explored to study the prevalence and relevance of such kinases to achieve better understanding the finer level functions of protein kinase sequences and their total functional space in regulating signal transduction pathways. Discovery of more domain families and better methods to designate domains to sequences would aid in further characterizing the expanding repertoire of kinases.
Acknowledgements
This research is supported by Department of Biotechnology, Government of India as well as by the Mathematical Biology Program, Department of Science and Technology, Government of India. MM is supported by Kothari fellowship, University Grants Commission. NS is a J C Bose National Fellow.References
- R. L. Marsden, D. Lee, M. Maibaum, C. Yeats and C. A. Orengo, Nucleic Acids Res., 2006, 34, 1066–1080 CrossRef CAS PubMed.
- C. Vogel, M. Bashton, N. D. Kerrison, C. Chothia and S. A. Teichmann, Curr. Opin. Struct. Biol., 2004, 14, 208–216 CrossRef CAS PubMed.
- G. Manning, WormBook, 2005 Search PubMed.
- P. Lahiry, A. Torkamani, N. J. Schork and R. A. Hegele, Nat. Rev. Genet., 2010, 11, 60–74 CrossRef CAS PubMed.
- M. Rask-Andersen, M. S. Almén and H. B. Schiöth, Nat. Rev. Drug Discovery, 2011, 10, 579–590 CrossRef CAS PubMed.
- J. A. Ubersax and J. E. Ferrell, Nat. Rev. Mol. Cell Biol., 2007, 8, 530–541 CrossRef CAS PubMed.
- S. K. Hanks and T. Hunter, FASEB J., 1995, 9, 576–596 CAS.
- R. Rakshambikai, M. Gnanavel and N. Srinivasan, PLoS One, 2014, 9, e107956 Search PubMed.
- K. Deshmukh, K. Anamika and N. Srinivasan, Prog. Biophys. Mol. Biol., 2010, 102, 1–15 CrossRef CAS PubMed.
- R. P. Bhattacharyya, A. Reményi, B. J. Yeh and W. A. Lim, Annu. Rev. Biochem., 2006, 75, 655–680 CrossRef CAS PubMed.
- G. Manning, D. B. Whyte, R. Martinez, T. Hunter and S. Sudarsanam, Science, 2002, 298, 1912–1934 CrossRef CAS PubMed.
- J. M. Arencibia, D. Pastor-Flores, A. F. Bauer, J. O. Schulze and R. M. Biondi, Biochim. Biophys. Acta, 2013, 1834, 1302–1321 CrossRef CAS PubMed.
- R. A. Currie, K. S. Walker, A. Gray, M. Deak, A. Casamayor, C. P. Downes, P. Cohen, D. R. Alessi and J. Lucocq, Biochem. J., 1999, 337(3), 575–583 CrossRef CAS.
- M. Tessier and J. R. Woodgett, J. Biol. Chem., 2006, 281, 23978–23989 CrossRef CAS PubMed.
- A. C. Newton, Biochem. J., 2003, 370, 361–371 CrossRef CAS PubMed.
- P. Flynn, H. Mellor, A. Casamassima and P. J. Parker, J. Biol. Chem., 2000, 275, 11064–11070 CrossRef CAS PubMed.
- R. Anjum and J. Blenis, Nat. Rev. Mol. Cell Biol., 2008, 9, 747–758 CrossRef CAS PubMed.
- J. A. Pitcher, N. J. Freedman and R. J. Lefkowitz, Annu. Rev. Biochem., 1998, 67, 653–692 CrossRef CAS PubMed.
- M. Amano, Y. Fukata and K. Kaibuchi, Exp. Cell Res., 2000, 261, 44–51 CrossRef CAS PubMed.
- M. Valiente, A. Andrés-Pons, B. Gomar, J. Torres, A. Gil, C. Tapparel, S. E. Antonarakis and R. Pulido, J. Biol. Chem., 2005, 280, 28936–28943 CrossRef CAS PubMed.
- S. Bialik and A. Kimchi, Annu. Rev. Biochem., 2006, 75, 189–210 CrossRef CAS PubMed.
- T. Tanaka, H. Koizumi and J. G. Gleeson, Cereb. Cortex, 2006, 16(suppl. 1), i69–i73 CrossRef PubMed.
- L. Funke, S. Dakoji and D. S. Bredt, Annu. Rev. Biochem., 2005, 74, 219–245 CrossRef CAS PubMed.
- A. R. Cohen, D. F. Woods, S. M. Marfatia, Z. Walther, A. H. Chishti, J. M. Anderson and D. F. Wood, J. Cell Biol., 1998, 142, 129–138 CrossRef CAS.
- A. Debant, C. Serra-Pagès, K. Seipel, S. O'Brien, M. Tang, S. H. Park and M. Streuli, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 5466–5471 CrossRef CAS.
- O. S. Rosenberg, S. Deindl, R.-J. Sung, A. C. Nairn and J. Kuriyan, Cell, 2005, 123, 849–860 CrossRef CAS PubMed.
- T. Iglesias and E. Rozengurt, J. Biol. Chem., 1998, 273, 410–416 CrossRef CAS PubMed.
- Z. Sun, J. Hsiao, D. S. Fay and D. F. Stern, Science, 1998, 281, 272–274 CrossRef CAS.
- K. Kohama, T. Okagaki, K. Hayakawa, Y. Lin, R. Ishikawa, T. Shimmen and A. Inoue, Biochem. Biophys. Res. Commun., 1992, 184, 1204–1211 CrossRef CAS.
- L. C. Meyer and N. T. Wright, Front. Physiol., 2013, 4, 1–12 Search PubMed.
- U. Knippschild, A. Gocht, S. Wolff, N. Huber, J. Löhler and M. Stöter, Cell. Signalling, 2005, 17, 675–689 CrossRef CAS PubMed.
- N. Kannan and A. F. Neuwald, Protein Sci., 2004, 13, 2059–2077 CrossRef CAS PubMed.
- P. Blume-Jensen and T. Hunter, Nature, 2001, 411, 355–365 CrossRef CAS PubMed.
- W. T. Miller, Acc. Chem. Res., 2003, 36, 393–400 CrossRef CAS PubMed.
- M. T. Brown and J. A. Cooper, Biochim. Biophys. Acta, 1996, 1287, 121–149 Search PubMed.
- A. M. Pendergast, Adv. Cancer Res., 2002, 85, 51–100 CrossRef CAS.
- J. M. Dunty, V. Gabarra-Niecko, M. L. King, D. F. J. Ceccarelli, M. J. Eck and M. D. Schaller, Mol. Cell. Biol., 2004, 24, 5353–5368 CrossRef CAS PubMed.
- S. J. Isakoff, T. Cardozo, J. Andreev, Z. Li, K. M. Ferguson, R. Abagyan, M. A. Lemmon, A. Aronheim and E. Y. Skolnik, EMBO J., 1998, 17, 5374–5387 CrossRef CAS PubMed.
- V. A. McPherson, S. Everingham, R. Karisch, J. A. Smith, C. M. Udell, J. Zheng, Z. Jia and A. W. B. Craig, Mol. Cell. Biol., 2009, 29, 389–401 CrossRef CAS PubMed.
- D. G. Woodside, A. Obergfell, A. Talapatra, D. A. Calderwood, S. J. Shattil and M. H. Ginsberg, J. Biol. Chem., 2002, 277, 39401–39408 CrossRef CAS PubMed.
- I. N. Maruyama, Cells, 2014, 3, 304–330 CrossRef CAS PubMed.
- D. L. Wheeler and Y. Yarden, Receptor Tyrosine Kinases: Structure, Functions and Role in Human Disease, Springer, 2014 Search PubMed.
- C. Wiesmann, Y. A. Muller and A. M. de Vos, J. Mol. Med., 2000, 78, 247–260 CrossRef CAS.
- J.-P. Himanen, N. Saha and D. B. Nikolov, Curr. Opin. Cell Biol., 2007, 19, 534–542 CrossRef CAS PubMed.
- H. Xu, D. Bihan, F. Chang, P. H. Huang, R. W. Farndale and B. Leitinger, PLoS One, 2012, 7, e52209 CAS.
- C. E. Lorén, C. Englund, C. Grabbe, B. Hallberg, T. Hunter and R. H. Palmer, EMBO Rep., 2003, 4, 781–786 CrossRef PubMed.
- M. Nomi, I. Oishi, S. Kani, H. Suzuki, T. Matsuda, A. Yoda, M. Kitamura, K. Itoh, S. Takeuchi, K. Takeda, S. Akira, M. Ikeya, S. Takada and Y. Minami, Mol. Cell. Biol., 2001, 21, 8329–8335 CrossRef CAS PubMed.
- R. van Amerongen and R. Nusse, Development, 2009, 136, 3205–3214 CrossRef CAS PubMed.
- M. E. Pitulescu and R. H. Adams, Genes Dev., 2010, 24, 2480–2492 CrossRef CAS PubMed.
- D. Neumann, C. Kollewe, A. Pich, P. Cao, K. Resch and M. U. Martin, J. Leukocyte Biol., 2008, 84, 807–813 CrossRef CAS PubMed.
- K. Mizuno, I. Okano, K. Ohashi, K. Nunoue, K. Kuma, T. Miyata and T. Nakamura, Oncogene, 1994, 9, 1605–1612 CAS.
- J.-M. Taymans, Biochem. Soc. Trans., 2012, 40, 1063–1069 CrossRef CAS PubMed.
- R. K. Rasmussen, J. Rusak, G. Price, P. J. Robinson, R. J. Simpson and D. S. Dorow, Biochem. J., 1998, 335(1), 119–124 CAS.
- N. H. Tran, X. Wu and J. A. Frost, J. Biol. Chem., 2005, 280, 16244–16253 CrossRef CAS PubMed.
- A. S. Dhillon, S. Meikle, Z. Yazici, M. Eulitz and W. Kolch, EMBO J., 2002, 21, 64–71 CrossRef CAS.
- N. Josso and N. di Clemente, Curr. Opin. Genet. Dev., 1997, 7, 371–377 CrossRef CAS.
- L. F. Andrade, L. A. Nahum, L. G. Avelar, L. L. Silva, A. Zerlotini, J. C. Ruiz and G. Oliveira, BMC Genomics, 2011, 12, 215 CrossRef CAS PubMed.
- F. Villa, J. Goebel, F. H. Rafiqi, M. Deak, J. Thastrup, D. R. Alessi and D. M. F. van Aalten, EMBO Rep., 2007, 8, 839–845 CrossRef CAS PubMed.
- J. D. Carpten, A. L. Faber, C. Horn, G. P. Donoho, S. L. Briggs, C. M. Robbins, G. Hostetter, S. Boguslawski, T. Y. Moses, S. Savage, M. Uhlik, A. Lin, J. Du, Y.-W. Qian, D. J. Zeckner, G. Tucker-Kellogg, J. Touchman, K. Patel, S. Mousses, M. Bittner, R. Schevitz, M.-H. T. Lai, K. L. Blanchard and J. E. Thomas, Nature, 2007, 448, 439–444 CrossRef CAS PubMed.
- M. J. Lindhurst, J. C. Sapp, J. K. Teer, J. J. Johnston, E. M. Finn, K. Peters, J. Turner, J. L. Cannons, D. Bick, L. Blakemore, C. Blumhorst, K. Brockmann, P. Calder, N. Cherman, M. A. Deardorff, D. B. Everman, G. Golas, R. M. Greenstein, B. M. Kato, K. M. Keppler-Noreuil, S. A. Kuznetsov, R. T. Miyamoto, K. Newman, D. Ng, K. O'Brien, S. Rothenberg, D. J. Schwartzentruber, V. Singhal, R. Tirabosco, J. Upton, S. Wientroub, E. H. Zackai, K. Hoag, T. Whitewood-Neal, P. G. Robey, P. L. Schwartzberg, T. N. Darling, L. L. Tosi, J. C. Mullikin and L. G. Biesecker, N. Engl. J. Med., 2011, 365, 611–619 CrossRef CAS PubMed.
- K. Hussain, B. Challis, N. Rocha, F. Payne, M. Minic, A. Thompson, A. Daly, C. Scott, J. Harris, B. J. L. Smillie, D. B. Savage, U. Ramaswami, P. De Lonlay, S. O'Rahilly, I. Barroso and R. K. Semple, Science, 2011, 334, 474 CrossRef CAS PubMed.
- J. H. Lee, M. Huynh, J. L. Silhavy, S. Kim, T. Dixon-Salazar, A. Heiberg, E. Scott, V. Bafna, K. J. Hill, A. Collazo, V. Funari, C. Russ, S. B. Gabriel, G. W. Mathern and J. G. Gleeson, Nat. Genet., 2012, 44, 941–945 CrossRef CAS PubMed.
- A. Poduri, G. D. Evrony, X. Cai, P. C. Elhosary, R. Beroukhim, M. K. Lehtinen, L. B. Hills, E. L. Heinzen, A. Hill, R. S. Hill, B. J. Barry, B. F. D. Bourgeois, J. J. Riviello, A. J. Barkovich, P. M. Black, K. L. Ligon and C. A. Walsh, Neuron, 2012, 74, 41–48 CrossRef CAS PubMed.
- H. Morita, K. Yoshida, K. Suzuki and S. Ikeda, J. Hum. Genet., 2006, 51, 1118–1121 CrossRef PubMed.
- I. Yabe, H. Sasaki, D.-H. Chen, W. H. Raskind, T. D. Bird, I. Yamashita, S. Tsuji, S. Kikuchi and K. Tashiro, Arch. Neurol., 2003, 60, 1749–1751 CrossRef PubMed.
- D.-H. Chen, Z. Brkanac, C. L. M. J. Verlinde, X.-J. Tan, L. Bylenok, D. Nochlin, M. Matsushita, H. Lipe, J. Wolff, M. Fernandez, P. J. Cimino, T. D. Bird and W. H. Raskind, Am. J. Hum. Genet., 2003, 72, 839–849 CrossRef CAS.
- D. S. Verbeek, M. A. Knight, G. G. Harmison, K. H. Fischbeck and B. W. Howell, Brain J. Neurol., 2005, 128, 436–442 CrossRef CAS PubMed.
- Z. Brkanac, L. Bylenok, M. Fernandez, M. Matsushita, H. Lipe, J. Wolff, D. Nochlin, W. H. Raskind and T. D. Bird, Arch. Neurol., 2002, 59, 1291–1295 CrossRef.
- V. Tran-Fadulu, J. H. Chen, D. Lemuth, B. T. Neichoy, J. Yuan, N. Gomes, E. Sparks, L. A. Kramer, D. Guo, H. Pannu, A. C. Braverman, S. Shete and D. M. Milewicz, Am. J. Med. Genet., Part A, 2006, 140, 1196–1202 CrossRef PubMed.
- D. Guo, E. Regalado, D. E. Casteel, R. L. Santos-Cortez, L. Gong, J. J. Kim, S. Dyack, S. G. Horne, G. Chang, G. Jondeau, C. Boileau, J. S. Coselli, Z. Li, S. M. Leal, J. Shendure, M. J. Rieder, M. J. Bamshad, D. A. Nickerson, GenTAC Registry Consortium, National Heart, Lung, and Blood Institute Grand Opportunity Exome Sequencing Project, C. Kim and D. M. Milewicz, Am. J. Hum. Genet., 2013, 93, 398–404 CrossRef CAS PubMed.
- H. Lee, J. M. Graham, D. L. Rimoin, R. S. Lachman, P. Krejci, S. W. Tompson, S. F. Nelson, D. Krakow and D. H. Cohn, Am. J. Hum. Genet., 2012, 90, 746–751 CrossRef CAS PubMed.
- M. Itoh-Satoh, T. Hayashi, H. Nishi, Y. Koga, T. Arimura, T. Koyanagi, M. Takahashi, S. Hohda, K. Ueda, T. Nouchi, M. Hiroe, F. Marumo, T. Imaizumi, M. Yasunami and A. Kimura, Biochem. Biophys. Res. Commun., 2002, 291, 385–393 CrossRef CAS PubMed.
- P. B. Agrawal, C. R. Pierson, M. Joshi, X. Liu, G. Ravenscroft, B. Moghadaszadeh, T. Talabere, M. Viola, L. C. Swanson, G. Haliloğlu, B. Talim, K. S. Yau, R. J. N. Allcock, N. G. Laing, M. A. Perrella and A. H. Beggs, Am. J. Hum. Genet., 2014, 95, 218–226 CrossRef CAS PubMed.
- L. Wang, D. Guo, J. Cao, L. Gong, K. E. Kamm, E. Regalado, L. Li, S. Shete, W.-Q. He, M.-S. Zhu, S. Offermanns, D. Gilchrist, J. Elefteriades, J. T. Stull and D. M. Milewicz, Am. J. Hum. Genet., 2010, 87, 701–707 CrossRef CAS PubMed.
- C. Cybulski, T. Huzarski, B. Górski, B. Masojć, M. Mierzejewski, T. Debniak, B. Gliniewicz, J. Matyjasik, E. Złowocka, G. Kurzawski, A. Sikorski, M. Posmyk, M. Szwiec, R. Czajka, S. A. Narod and J. Lubiński, Cancer Res., 2004, 64, 2677–2679 CrossRef CAS.
- G. Piluso, F. D'Amico, V. Saccone, E. Bismuto, I. L. Rotundo, M. Di Domenico, S. Aurino, C. E. Schwartz, G. Neri and V. Nigro, Am. J. Hum. Genet., 2009, 84, 162–177 CrossRef CAS PubMed.
- R. Bocciardi, D. Bordo, M. Di Duca, M. Di Rocco and R. Ravazzolo, Eur. J. Hum. Genet., 2009, 17, 311–318 CAS.
- G.-T. Lin, H.-W. Chang, C.-S. Liu, P.-J. Huang, H.-C. Wang and Y.-M. Cheng, J. Hum. Genet., 2006, 51, 1083–1086 CrossRef PubMed.
- M. Nakajima, N. Haga, K. Takikawa, N. Manabe, G. Nishimura and S. Ikegawa, J. Hum. Genet., 2007, 52, 473–475 CrossRef PubMed.
- E. M. Shore, M. Xu, G. J. Feldman, D. A. Fenstermacher, T.-J. Cho, I. H. Choi, J. M. Connor, P. Delai, D. L. Glaser, M. LeMerrer, R. Morhart, J. G. Rogers, R. Smith, J. T. Triffitt, J. A. Urtizberea, M. Zasloff, M. A. Brown and F. S. Kaplan, Nat. Genet., 2006, 38, 525–527 CrossRef CAS PubMed.
- K. A. Petrie, W. H. Lee, A. N. Bullock, J. J. Pointon, R. Smith, R. G. G. Russell, M. A. Brown, B. P. Wordsworth and J. T. Triffitt, PLoS One, 2009, 4, e5005 Search PubMed.
- B. Pandit, A. Sarkozy, L. A. Pennacchio, C. Carta, K. Oishi, S. Martinelli, E. A. Pogna, W. Schackwitz, A. Ustaszewska, A. Landstrom, J. M. Bos, S. R. Ommen, G. Esposito, F. Lepri, C. Faul, P. Mundel, J. P. López Siguero, R. Tenconi, A. Selicorni, C. Rossi, L. Mazzanti, I. Torrente, B. Marino, M. C. Digilio, G. Zampino, M. J. Ackerman, B. Dallapiccola, M. Tartaglia and B. D. Gelb, Nat. Genet., 2007, 39, 1007–1012 CrossRef CAS PubMed.
- M. A. Razzaque, T. Nishizawa, Y. Komoike, H. Yagi, M. Furutani, R. Amo, M. Kamisago, K. Momma, H. Katayama, M. Nakagawa, Y. Fujiwara, M. Matsushima, K. Mizuno, M. Tokuyama, H. Hirota, J. Muneuchi, T. Higashinakagawa and R. Matsuoka, Nat. Genet., 2007, 39, 1013–1017 CrossRef CAS PubMed.
- P. S. Dhandapany, M. A. Razzaque, U. Muthusami, S. Kunnoth, J. J. Edwards, S. Mulero-Navarro, I. Riess, S. Pardo, J. Sheng, D. S. Rani, B. Rani, P. Govindaraj, E. Flex, T. Yokota, M. Furutani, T. Nishizawa, T. Nakanishi, J. Robbins, G. Limongelli, R. J. Hajjar, D. Lebeche, A. Bahl, M. Khullar, A. Rathinavel, K. C. Sadler, M. Tartaglia, R. Matsuoka, K. Thangaraj and B. D. Gelb, Nat. Genet., 2014, 46, 635–639 CrossRef CAS PubMed.
- T. Schindler, W. Bornmann, P. Pellicena, W. T. Miller, B. Clarkson and J. Kuriyan, Science, 2000, 289, 1938–1942 CrossRef CAS.
- D. C. Saffran, O. Parolini, M. E. Fitch-Hilgenberg, D. J. Rawlings, D. E. Afar, O. N. Witte and M. E. Conley, N. Engl. J. Med., 1994, 330, 1488–1491 CrossRef CAS PubMed.
- D. J. Rawlings and O. N. Witte, Immunol. Rev., 1994, 138, 105–119 CrossRef CAS PubMed.
- Q. Zhu, M. Zhang, J. Winkelstein, S. H. Chen and H. D. Ochs, Hum. Mol. Genet., 1994, 3, 1899–1900 CrossRef CAS PubMed.
- M. E. Conley and J. M. Puck, J. Pediatr., 1988, 112, 688–694 CrossRef CAS.
- B. R. Ali, H. Xu, N. A. Akawi, A. John, N. S. Karuvantevida, R. Langer, L. Al-Gazali and B. Leitinger, Hum. Mol. Genet., 2010, 19, 2239–2250 CrossRef CAS PubMed.
- A. Shiels, T. M. Bennett, H. L. S. Knopf, G. Maraini, A. Li, X. Jiao and J. F. Hejtmancik, Mol. Vision, 2008, 14, 2042–2055 CAS.
- T. Zhang, R. Hua, W. Xiao, K. P. Burdon, S. S. Bhattacharya, J. E. Craig, D. Shang, X. Zhao, D. A. Mackey, A. T. Moore, Y. Luo, J. Zhang and X. Zhang, Hum. Mutat., 2009, 30, E603–E611 CrossRef PubMed.
- M. R. Passos-Bueno, W. R. Wilcox, E. W. Jabs, A. L. Sertié, L. G. Alonso and H. Kitoh, Hum. Mutat., 1999, 14, 115–125 CrossRef CAS.
- N. Pitteloud, J. S. Acierno, A. Meysing, A. V. Eliseenkova, J. Ma, O. A. Ibrahimi, D. L. Metzger, F. J. Hayes, A. A. Dwyer, V. A. Hughes, M. Yialamas, J. E. Hall, E. Grant, M. Mohammadi and W. F. Crowley, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 6281–6286 CrossRef CAS PubMed.
- E. B. Trarbach, E. M. F. Costa, B. Versiani, M. de Castro, M. T. M. Baptista, H. M. Garmes, B. B. de Mendonca and A. C. Latronico, J. Clin. Endocrinol. Metab., 2006, 91, 4006–4012 CrossRef CAS PubMed.
- H. Miraoui, A. A. Dwyer, G. P. Sykiotis, L. Plummer, W. Chung, B. Feng, A. Beenken, J. Clarke, T. H. Pers, P. Dworzynski, K. Keefe, M. Niedziela, T. Raivio, W. F. Crowley, S. B. Seminara, R. Quinton, V. A. Hughes, P. Kumanov, J. Young, M. A. Yialamas, J. E. Hall, G. Van Vliet, J.-P. Chanoine, J. Rubenstein, M. Mohammadi, P.-S. Tsai, Y. Sidis, K. Lage and N. Pitteloud, Am. J. Hum. Genet., 2013, 92, 725–743 CrossRef CAS PubMed.
- D. Steinberger, J. B. Mulliken and U. Müller, Hum. Genet., 1995, 96, 113–115 CrossRef CAS.
- W. Reardon, R. M. Winter, P. Rutland, L. J. Pulleyn, B. M. Jones and S. Malcolm, Nat. Genet., 1994, 8, 98–103 CrossRef CAS PubMed.
- S. Heuertz, M. Le Merrer, B. Zabel, M. Wright, L. Legeai-Mallet, V. Cormier-Daire, L. Gibbs and J. Bonaventure, Eur. J. Hum. Genet., 2006, 14, 1240–1247 CAS.
- A. Ghalamkarpour, S. Morlot, A. Raas-Rothschild, A. Utkus, J. B. Mulliken, L. M. Boon and M. Vikkula, Clin. Genet., 2006, 70, 330–335 CrossRef CAS PubMed.
- K. Huck, O. Feyen, T. Niehues, F. Rüschendorf, N. Hübner, H.-J. Laws, T. Telieps, S. Knapp, H.-H. Wacker, A. Meindl, H. Jumaa and A. Borkhardt, J. Clin. Invest., 2009, 119, 1350–1358 CrossRef CAS.
- R. M. Linka, S. L. Risse, K. Bienemann, M. Werner, Y. Linka, F. Krux, C. Synaeve, R. Deenen, S. Ginzel, R. Dvorsky, M. Gombert, A. Halenius, R. Hartig, M. Helminen, A. Fischer, P. Stepensky, K. Vettenranta, K. Köhrer, M. R. Ahmadian, H.-J. Laws, B. Fleckenstein, H. Jumaa, S. Latour, B. Schraven and A. Borkhardt, Leukemia, 2012, 26, 963–971 CrossRef CAS PubMed.
- K. Touhara, W. J. Koch, B. E. Hawes and R. J. Lefkowitz, J. Biol. Chem., 1995, 270, 17000–17005 CrossRef CAS PubMed.
- P. Penela, C. Ribas and F. Mayor, Cell. Signalling, 2003, 15, 973–981 CrossRef CAS.
- M. Jaleel, F. Villa, M. Deak, R. Toth, A. R. Prescott, D. M. F. van Aalten and D. R. Alessi, Biochem. J., 2006, 394, 545–555 CrossRef CAS PubMed.
- N. Yabuta, S. Mukai, A. Okamoto, D. Okuzaki, H. Suzuki, K. Torigata, K. Yoshida, N. Okada, D. Miura, A. Ito, M. Ikawa, M. Okabe and H. Nojima, J. Cell Sci., 2013, 126, 508–520 CrossRef CAS PubMed.
- H. A. Burgess and O. Reiner, J. Biol. Chem., 2001, 276, 36397–36403 CrossRef CAS PubMed.
- D. Koveal, N. Schuh-Nuhfer, D. Ritt, R. Page, D. K. Morrison and W. Peti, Sci. Signaling, 2012, 5, ra94 CrossRef PubMed.
- D. Buck, J. E. Smith, C. S. Chung, Y. Ono, H. Sorimachi, S. Labeit and H. L. Granzier, J. Gen. Physiol., 2014, 143, 215–230 CrossRef CAS PubMed.
- N. Mizushima, T. Noda and Y. Ohsumi, EMBO J., 1999, 18, 3888–3896 CrossRef CAS PubMed.
- M. Matsushita, N. N. Suzuki, Y. Fujioka, Y. Ohsumi and F. Inagaki, Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun., 2006, 62, 1021–1023 CrossRef CAS PubMed.
- K. L. Borden and P. S. Freemont, Curr. Opin. Struct. Biol., 1996, 6, 395–401 CrossRef CAS.
- A. Hausser, P. Storz, S. Hübner, I. Braendlin, M. Martinez-Moya, G. Link and F.-J. Johannes, FEBS Lett., 2001, 492, 39–44 CrossRef CAS.
- J. S. Michaelson, Apoptosis, 2000, 5, 217–220 CrossRef CAS.
- C. Martín-Castellanos, M. Blanco, A. E. Rozalén, L. Pérez-Hidalgo, A. I. García, F. Conde, J. Mata, C. Ellermeier, L. Davis, P. San-Segundo, G. R. Smith and S. Moreno, Curr. Biol., 2005, 15, 2056–2062 CrossRef PubMed.
- T. Hubbard, D. Barker, E. Birney, G. Cameron, Y. Chen, L. Clark, T. Cox, J. Cuff, V. Curwen, T. Down, R. Durbin, E. Eyras, J. Gilbert, M. Hammond, L. Huminiecki, A. Kasprzyk, H. Lehvaslaiho, P. Lijnzaad, C. Melsopp, E. Mongin, R. Pettett, M. Pocock, S. Potter, A. Rust, E. Schmidt, S. Searle, G. Slater, J. Smith, W. Spooner, A. Stabenau, J. Stalker, E. Stupka, A. Ureta-Vidal, I. Vastrik and M. Clamp, Nucleic Acids Res., 2002, 30, 38–41 CrossRef CAS.
Footnotes |
| † Joint first authors. |
| ‡ Present address: Molecular Signaling Lab, Signal Processing Department, Tampere University of Technology, Tampere Finland. |
| This journal is © The Royal Society of Chemistry 2015 |