
Hydrolase-catalyzed asymmetric carbon–carbon bond formation in organic synthesis
Zhi Guan
*,
Ling-Yu Li
and
Yan-Hong He
*
Key Laboratory of Applied Chemistry of Chongqing Municipality, School of Chemistry and Chemical Engineering, Southwest University, Chongqing 400715, PR China. E-mail: guanzhi@swu.edu.cn; heyh@swu.edu.cn; Fax: +86-23-68254091
First published on 15th January 2015
Abstract
Enzyme catalytic promiscuity, in which the active site of an enzyme has the ability to catalyze more than one chemical transformation, has received widespread attention as more catalytic promiscuities of existing enzymes have been discovered. In this field, hydrolases have been mainly studied due to their commercial availability, high stability, broad substrate scope and high catalytic efficiency in media containing organic solvents. In this study, we review the hydrolase-catalyzed asymmetric carbon–carbon bond-forming reactions for the preparation of enantiomerically enriched compounds in organic synthesis. To date, these hydrolase-catalyzed asymmetric reactions include the direct asymmetric aldol, Michael, Mannich and Morita–Baylis–Hillman reactions. The hydrolase-catalyzed non-enantioselective examples were not included.
 Zhi Guan | Zhi Guan was born in Yunnan, China, in 1964. He received his B.S. in Chemistry from Yunnan University in 1988. He got his PhD in 2000 from Sun Yat-sen University under the guidance of Prof. Long-Mei Zeng. He pursued postdoctoral studies with Prof. Xiao-Dong Su at the Dept. of Molecular Biophysics, Lund University, Sweden; Prof. Adolf Gogoll at Dept. of Organic Chemistry, Uppsala University, Sweden; and Dr Philip Lowden at the University of London, UK. In 2005 he became an associate professor of organic chemistry at Southwest University, China. His current research interests include biocatalysis and organic synthesis. |
 Ling-Yu Li | Ling-Yu Li was born in Cheng Du, China, in 1988. She received her B.S. degree in Applied Chemistry from Mianyang Normal University in 2012. Currently, she is studying for a Master Degree in Organic Chemistry under the supervision of Dr Yan-Hong He at Southwest University. She has investigated some asymmetric organic reactions catalyzed by promiscuous enzymes. Her research interests are focused on organic synthesis and bioorganic chemistry. |
 Yan-Hong He | Yan-Hong He was born in Yunnan, China, in 1966. She received her B.S. in Chemistry from Yunnan University in 1988. During 1988–1996, she was a research fellow in Yunnan Tropical Crops Research Institute. She completed her M.S. under the supervision of Prof. Long-Mei Zeng in 1999 at Sun Yat-sen University. During 1999–2001, she was a lecturer at Sun Yat-sen University. She got her PhD under the supervision of Prof. Olov Sterner in 2004 at the Dept. of Organic and Bioorganic Chemistry, Lund University, Sweden. In 2005 she became an associate professor at Southwest University, China. Her research interests focus on organic synthesis and bioorganic chemistry. |
1. Introduction
Biocatalysis is the application of enzymes for the chemical transformation of organic compounds.1 The advent of biocatalysis as an elegant synthetic methodology meets the development of sustainable chemistry.2 The enzymatic process as a widely used biocatalysis technology is growing increasingly in the field of catalysis. In metabolism, an enzyme is evolutionarily optimized through recognizing a specific substrate for a specific transformation.1,3 In spite of that many enzymes can also show promiscuous behavior, which is often hidden behind a native catalytic transformation.1,4–6 In many cases, these activities for promiscuous substrates are usually very low in comparison with their native activities and hence might have slight physiological relevance.7 However, promiscuous activities are useful for organic synthesis. Therefore, the research on enzyme promiscuity not only gains insight on the evolution of new enzymes, but also expands the application of biocatalysts in organic synthesis. According to Hult and Berglund, enzyme promiscuity can be divided into three main aspects: (1) condition promiscuity, meaning that the activity of the enzyme persists under reaction conditions that are not natural; (2) substrate promiscuity, meaning that an enzyme can tolerate a wide range of substrates; (3) catalytic promiscuity, meaning that the active site of an enzyme has the ability to catalyze more than one chemical transformation.3 This review is focused on catalytic promiscuity. In recent years, more and more enzymes have been found to display catalytic promiscuity for organic transformations. In this field, hydrolases have been mainly studied due to their commercial availability, high stability, broad substrate scope and high catalytic efficiency in media containing organic solvents. Hydrolases, a class of enzymes whose natural function is catalyzing the cleavage of chemical bonds using water as a nucleophile, have also been applied to synthesize organic compounds through some other catalytic ways such as the formation of carbon–carbon and carbon-heteroatom bonds. Asymmetric carbon–carbon bond formation is a general, efficient and important approach for producing enantiopure or enantio-enriched compounds. There have been some successful examples of the use of catalytically promiscuous enzymes as catalysts for asymmetric carbon–carbon bond formations, and hydrolases particularly played a crucial role in this aspect.2,8–13 This work reviewed the journal studies (the patent studies were not examined) that we found to date about hydrolase-catalyzed asymmetric carbon–carbon bond-forming reactions for the preparation of enantiomerically enriched compounds in organic synthesis, including direct asymmetric aldol, Michael, Mannich and Morita–Baylis–Hillman reactions.2. The hydrolase-catalyzed direct asymmetric aldol reactions
The aldol reaction is an important approach for building carbon–carbon bonds in organic chemistry and is often a critical step in overall synthesis.14–16 It may be characterized as a natural synthetic method because some substances of organism metabolism such as partial carbohydrates are based on the aldol reaction.17–19 Aldol reaction is relatively widely applied to either bulk production, or in the fine chemical and pharmaceutical industry.20 In addition to the aldol reactions naturally catalyzed by aldolases, some hydrolase-catalyzed aldol reactions have been successfully achieved.21,22 Hydrolases, such as lipases, proteases and nucleases, have been found to possess the ability to catalyze asymmetric aldol reactions. Herein, we want to highlight the hydrolase-catalyzed direct asymmetric aldol reactions.2.1. The lipase-catalyzed direct asymmetric aldol reactions
Lipases are one type of hydrolase that naturally catalyze the hydrolysis of long chain triglycerides and are also one class of the most widely used enzymes in organic synthesis.23 Some research groups have successfully applied lipases to catalyze carbon–carbon bond formations, including aldol reactions, Knoevenagel condensations, Michael additions, and Henry reactions.13,24,25 However, lipase-catalyzed carbon–carbon bond formation in an asymmetric manner is relatively few. In this section, the lipase-catalyzed direct asymmetric aldol reactions, reported in journals to date, are discussed according to different types of donors.
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Enzyme | Medium | Temp. (°C) | Time (h) | Yield (%) | ee (%) | Number of examples | Ref. |
| a PPL: lipase from porcine pancreas.b MJL: lipase from Mucor javanicus.c CAL-B: lipase from Candida antarctica.d MML: lipase from Mucor miehei.e ROL: lipase from Rhizopus oryzae.f BSL: Bacillus subtilis lipase.g PSL: Pseudomonas sp. lipase. | ||||||||
| 1 | PPLa | “Wet” acetone | 30 | 72–144 | Up to 96.4 | Up to 43.6 | 2 | 26 |
| 2 | MJLb | “Wet” acetone | 30 | 24 | 14.5 | 12.9 | 1 | 26 |
| 3 | CAL-Bc | “Wet” acetone | 30 | 24 | 2.3 | 9.4 | 1 | 26 |
| 4 | MMLd | “Wet” acetone | 30 | 24 | 9.8 | 9.6 | 1 | 26 |
| 5 | MML | Acetone/water | 30 | 24 | 9 | 9 | 1 | 27 |
| 6 | CAL-B | Acetone/water | 30 | 24 | 2 | 9 | 1 | 27 |
| 7 | ROLe | Acetone/water | 30 | 24 | 7 | 4 | 1 | 27 |
| 8 | MJL | Acetone/water | 30 | 24 | 15 | 12 | 1 | 27 |
| 9 | Lipase-AK | Acetone/water | 30 | 24 | 11 | 6 | 1 | 27 |
| 10 | PPL | Acetone/water | 37 | 72 | 57–64 | 17–23 | 2 | 28 |
| 11 | CAL-B | — | — | — | — | 10.5 | 1 | 29 |
| 12 | BSLf | — | — | — | — | 5.8 | 1 | 29 |
| 13 | PSLg | — | — | — | — | 16.8 | 1 | 29 |
| 14 | PPL | n-heptane/water | 30 | — | — | 46.9 | 1 | 29 |

 | ||
| Scheme 1 Proposed mechanism for the lipase-catalyzed aldol reaction. | ||
Subsequently, in 2010, Wang and co-workers assessed a wide variety of hydrolases for the aldol reaction of acetone with 4-nitrobenzaldehyde.27 However, the catalytic efficiency of lipases was not significantly improved (Table 1, entries 5–9). Later, in 2013, they also mentioned that acetone as the donor resulted in poor enantioselectivities using PPL in the presence of 5% water (Vwater/Vwater+acetone) (Table 1, entry 10).28 In 2014, catalytic promiscuity of hydrolase for the asymmetric aldol addition of 2-butanone and 4-nitrobenzaldehyde was examined by L. Wang and co-workers.29 Some lipases exhibited catalytic activities (Table 1, entries 11–14), and an ee of up to 46.9% was obtained using PPL as a catalyst in n-heptane with 8.4% water (Table 1, entry 14).
From the abovementioned examples of the lipase-catalyzed direct asymmetric aldol reactions of aromatic aldehydes with acyclic ketones, some critical information can be obtained as follows: some lipases have been discovered to possess the ability to catalyze the asymmetric aldol reactions of aldehydes with acyclic ketones; most of the listed lipases could maintain their catalytic activity in organic solvents in the presence of water; reaction conditions were mild with no need for high temperatures or special operations. However, quite low yields with poor enantioselectivities were obtained and long reaction times were often required.
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Enzyme | Medium | Temp. (°C) | Time (h) | Yield (%) | dr (anti/syn) | ee (%) | Number of examples | Ref. |
| a BPL: bovine pancreatic lipase.b PPL: lipase from porcine pancreas.c PPL II: porcine pancreas lipase type II.d ANL: amano lipase A from Aspergillus niger.e CCL: lipase from Candida cylindracea. | |||||||||
| 1 | BPLa | Buffer | 30 | 60–120 | 13–99 | 46/54–96/4 | 14–66 | 16 | 30 |
| 2 | BPL | Cyclohexanone/water | 37 | 24 | 86 | 81/19 | 79 | 1 | 28 |
| 3 | PPLb | Ketone/water | 37 | 30–120 | 70–99 | 47/53–99/1 | 32–90 | 16 | 28 |
| 4 | PPL IIc | MeCN/buffer | 20 | 144–192 | 10–98 | 48/52–87/13 | 53–94 | 15 | 31 |
| 5 | Lipase ps amano SD | MeCN/water | 25 | 168 | 5 | 57/43 | 48 | 1 | 31 |
| 6 | Lipase AK amano | MeCN/water | 25 | 168 | 10 | 67/33 | 30 | 1 | 31 |
| 7 | ANLd | MeCN/water | 25 | 168 | 25 | 57/43 | 54 | 1 | 31 |
| 8 | CCLe | MeCN/water | 25 | 168 | 10 | 74/26 | 40 | 1 | 31 |

In addition to BPL, PPL also showed excellent catalytic activity for the asymmetric aldol reactions of aromatic aldehydes with cyclic ketones. For instance, Wang et al. carried out the PPL-catalyzed aldol reactions of aromatic aldehydes and cyclic ketones in 2013.28 In their report, PPL was chosen as the best catalyst although BPL also gave reasonable results (Table 2, entry 2). The best results were obtained in wet cyclohexanone (5% water, by vol.). Control experiments indicated that the catalytic activity of PPL for the aldol reaction did not arise from nonspecific amino acids. The specific catalytic site and folding conformation of PPL were responsible for the aldol reaction. After optimizing the reaction conditions and substrate extension, products were achieved in desired yields with moderate to high enantioselectives (Table 2, entry 3). According to the abovementioned experiments, both PPL and BPL showed good catalytic effects in catalyzing the aldol reactions of aromatic aldehydes with cyclic ketones in terms of yields and enantioselectivies. This phenomenon probably could be attributed to the fact that the two pancreatic lipases possess a high degree of homology.
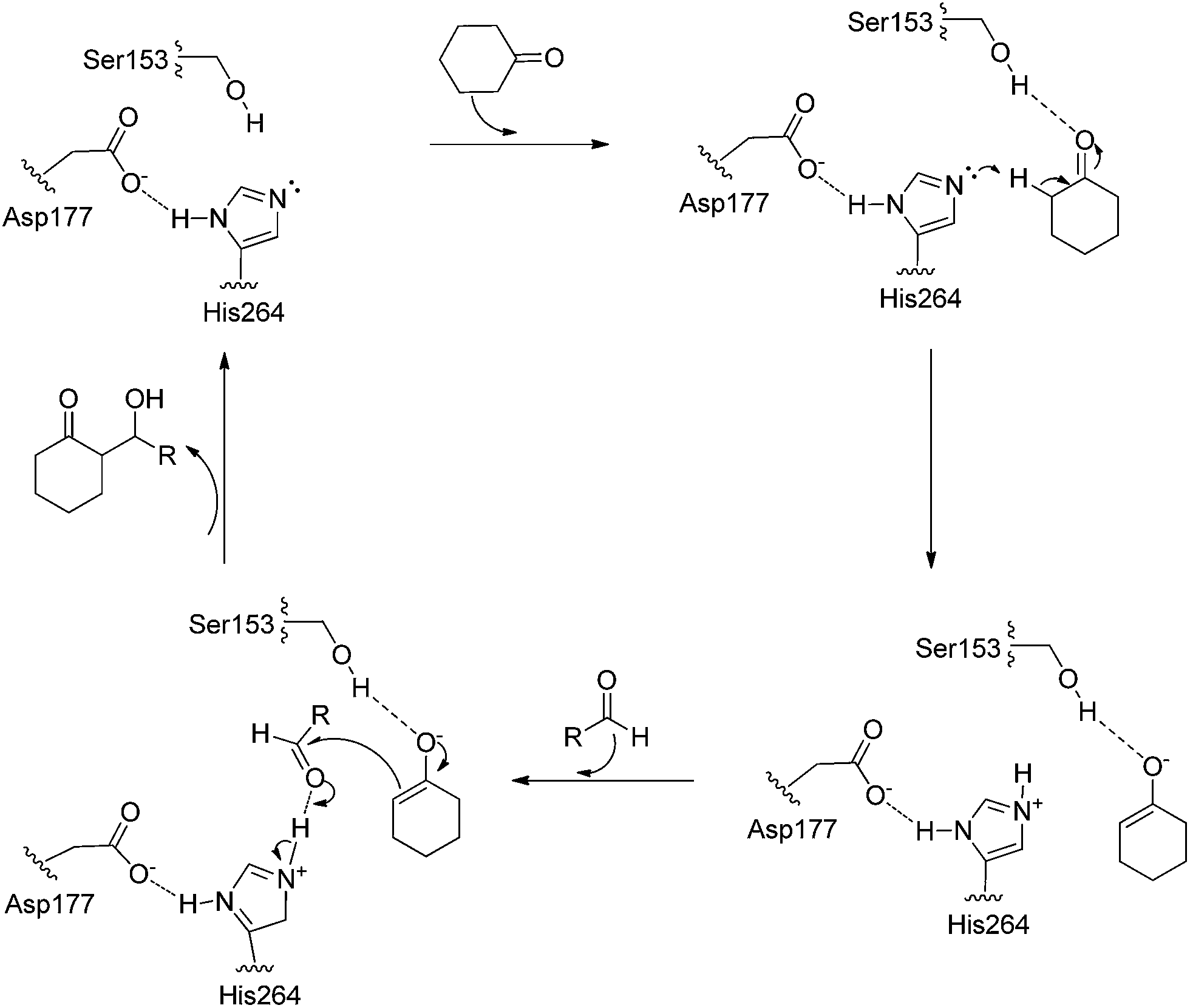
In addition, Wang et al. deduced a possible mechanism for the PPL-catalyzed asymmetric aldol reactions of aromatic aldehydes with cyclic ketones (Scheme 2).28 First, one of the substrates, cyclohexanone, is attached to the active site by the catalytic triad of Ser153, Asp177 and His264 in the active site. Then, an enolate anion transition state is formed through the transfer of a proton from cyclohexanone to His264. The enolate anion is stabilized by the Ser153 residue. After that, another substrate aldehyde captures the proton from the imidazolyl of His264 and interacts with the enolate anion to form a new carbon–carbon bond. Finally, the active site releases the aldol product and the catalytic process is finished.
 | ||
| Scheme 2 Proposed mechanism for the PPL-catalyzed aldol reaction. | ||
In 2014, our group discovered that some commercially available lipases could catalyze the direct asymmetric aldol reaction of cyclohexanone and 4-nitrobenzaldehyde (Table 2, entries 4–8).31 Among the tested enzymes, porcine pancreas lipase type II (PPL II) showed the highest catalytic activity and enantioselectivity in MeCN/buffer. A wide range of substrates were accepted by PPL II to afford the corresponding aldol products in low to high yields (10–98%) with moderate to excellent enantioselectivities (53–94% ee, for anti-isomers) and low to moderate diastereoselectivities (48/52–87/13 dr, anti/syn) (Table 2, entry 4).
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Enzyme | Medium | Temp. (°C) | Time (h) | Yield (%) | dr (anti/syn) | ee (%) | Number of examples | Ref. |
| a PPL II: lipase from porcine pancreas, type II.b WGL: lipase from wheat germ.c ANL-A: amano lipase A from Aspergillus niger. | |||||||||
| 1 | PPL IIa | MeCN/water | 30 | 120–144 | 31–56 | 32/68–83/17 | 43–87 | 12 | 35 |
| 2 | WGLb | MeCN/water | 25 | 120 | 28 | 54/46 | 12 | 1 | 35 |
| 3 | ANL-Ac | MeCN/water | 25 | 120 | 26 | 40/60 | 13 | 1 | 35 |

2.2. The protease-catalyzed direct asymmetric aldol reactions
Proteases, belonging to the class of hydrolases, catalyze the hydrolysis of peptide bonds in a general sense. Proteases are highly stereo- and region-specific catalysts, which are quite stable and easy to handle, usually act under mild reaction conditions, and are relatively simple in their molecular architecture in most cases. Historically, proteases have been associated with protein digestion in general.36 Since Klibanov et al. found that proteases can keep their activity for a long time in some organic solvents,37 the organic synthesis catalyzed by proteases began to be increasingly prosperous.38 In this section, the protease-catalyzed direct asymmetric aldol reactions reported in journals to date (to the best of our knowledge) are discussed.
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Enzyme | Medium | Temp. (°C) | Time (h) | Yield (%) | ee (%) | Ref. |
| a PPT: trypsin from porcine pancreas.b AMP: proteinase from Aspergillus melleus, type XXIII. | |||||||
| 1 | Pepsin | Acetone/water | 30 | 120 | 89 | 45 | 27 |
| 2 | Papain | Acetone/water | 30 | 24 | 18 | 11 | 27 |
| 3 | Trypsin | Acetone/water | 30 | 24 | 23 | 19 | 27 |
| 4 | Alcalase | Acetone/water | 45 | 48 | 68 | 13 | 39 |
| 5 | Chymopapain | MeCN/buffer | 30 | 120 | 12 | 14 | 40 |
| 6 | PPTa | Acetone/water | 30 | 46 | 28 | 16 | 41 |
| 7 | AMPb | MeCN/water | 30 | 192 | 20 | 32 | 19 |
In 2011, Gotor and co-workers examined different non-conventional hydrolase-catalyzed processes accelerated by protease from Bacillus licheniformis immobilized as cross-linked enzyme aggregates (alcalase-CLEA).39 They discovered that alcalase could promote the aldol transformation of 4-nitrobenzaldehyde and acetone, giving (R)-β-hydroxy ketone in a yield of 24% with 7% ee. A slight chiral induction toward the formation of the aldol product with 11% ee was observed when the reaction was performed in a wet environment. The effect of temperature was also explored under wet reaction conditions, and the best chiral induction (13% ee) with a higher reaction rate (68% yield) was observed when the temperature was controlled at 45 °C with 20% water content (Table 4, entry 4).
Our group also explored the asymmetric aldol reaction of 4-nitrobenzaldehyde and acetone in 2012 and 2013.19,40,41 Different catalytic activities were found in some proteases, including chymopapain, trypsin from porcine pancreas (PPT) and proteinase from Aspergillus melleus, type XXIII (AMP) (Table 4, entries 5–7). A small amount of water was essential for those proteases to maintain their catalytic activity in organic solvents, and almost all of the enzymatic reaction could be carried out at ambient temperature (around 30 °C). However, the investigated protease-catalyzed asymmetric aldol reaction of aromatic aldehydes with acyclic ketones was only limited to 4-nitrobenzaldehyde with acetone, and the reaction activities of other chain ketones need to be further examined.
In 2011, our group reported the BLAP-catalyzed enantioselective aldol additions of aromatic aldehydes and cyclic ketones,42 giving the corresponding products in yields from 28% to 92% with dr from 32/68 to 95/5 and ee from 22 to >99% in the solvent system of water/DMSO (0.15, v/v) (Table 5, entry 1). As a Ca2+-dependent enzyme, BLAP completely lost its catalytic activity for the aldol reaction after interacting with EDTA. Furthermore, the catalytic activity of BLAP was completely inhibited by the serine protease inhibitor, phenylmethanesulfonyl fluoride (PMSF), which demonstrated that the aldol reaction may occur in the active site of BLAP.
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Enzyme | Medium | Temp. (°C) | Time (h) | Yield (%) | dr (anti/syn) | ee (%) | Number of examples | Ref. |
| a BLAP: alkaline protease from Bacillus licheniformis.b AUAP: acidic protease from Aspergillus usamii.c PPT: trypsin from porcine pancreas.d AMP: proteinase from Aspergillus melleus. | |||||||||
| 1 | BLAPa | DMSO/water | 20 | 65–120 | 28–92 | 32/68–95/5 | 22–>99 | 13 | 42 |
| 2 | Chymopapain | MeCN/buffer | 30 | 120–240 | 12–73 | 47/53–>99/1 | 26–96 | 14 | 40 |
| 3 | AUAPb | MeCN/water | 25 | 144–168 | 10–78 | 53/47–97/3 | 52–88 | 16 | 43 |
| 4 | PPTc | Ketone/water | 30 | 71–192 | 7–60 | 11/89–89/11 | 33–65 | 11 | 41 |
| 5 | AMPd | MeCN/water | 30 | 96–216 | 11–89 | 47/53–>99/1 | 40–91 | 19 | 19 |
| 6 | Ficin | MeCN/water | 30 | 117 | 21–39 | 79/21–86/14 | 77–81 | 2 | 44 |

In 2012, we found that chymopapain, a cysteine proteinase isolated from the latex of the unripe fruits of Carica papaya, was a versatile catalyst for the aldol reactions of aromatic aldehydes with cyclic ketones in acetonitrile in the presence of phosphate buffer.40 Various products were obtained in low to moderate yields with up to >99/1 dr (anti/syn) and up to 96% ee (Table 5, entry 2). Based on the mechanism of the serine hydrolase CAL-B-catalyzed aldol reaction proposed by Berglund and co-workers,21 we speculated the mechanism of the chymopapain-catalyzed aldol reaction (Scheme 3).40 There is a catalytic triad formed by Cys, His and Asn, and an equilibrium exists between the neutral (thiol–imidazole) and the ion-pair (thiolate–imidazolium) of the Cys–His. First, the substrate ketone is grasped by the Asn–His dyad and the oxyanion hole in the active site through hydrogen bonding. Then, a proton on the ketone is removed by His residues, forming an enolate ion. Next, another substrate aldehyde acquires a proton from the protic imidazolium and then acts on the enolate ion forming a new carbon–carbon bond. Eventually, the target product is released from the oxyanion hole and is separated from the active site.
 | ||
| Scheme 3 Proposed mechanism for the chymopapain-catalyzed aldol reaction. | ||
The direct asymmetric aldol reactions catalyzed by another two proteases (AUAP and PPT) were also reported by our group in 2012.41,43 AUAP exhibited the preferable catalytic activity and stereoselectivity in MeCN in the presence of 10% water at 25 °C (Table 5, entry 3), whereas PPT showed its favorable catalytic ability to the asymmetric direct aldol reaction under organic solvent-free conditions with the ratio of enzyme to water of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.5 (enzyme–water, w/w) (Table 5, entry 4). Both AUAP and PPT could accept various aromatic aldehydes and cyclic ketones as substrates, although the yields and stereoselectivities were not very satisfactory. In addition, a tentative mechanism of the PPT-catalyzed aldol reaction of aldehyde with cyclohexanone was hypothesized (Scheme 4).41 Trypsin is one of the important serine proteases with its active center having a catalytic triad composed of His57, Asp102, and Ser195. Initially, the substrate cyclohexanone is stabilized through hydrogen bonding by the Asp–His dyad and the oxyanion hole which was formed by the backbone amide hydrogen atoms of Gly-193 and Ser-195 in the active site. Next, an enolate ion is formed after a proton on cyclohexanone is removed by the His residue. Then, a carbon–carbon bond is formed by the combination of the enolate ion with the aldehyde, which is protonated by the imidazolium cation. Eventually, the target aldol product is released from the oxyanion hole.
:1.5 (enzyme–water, w/w) (Table 5, entry 4). Both AUAP and PPT could accept various aromatic aldehydes and cyclic ketones as substrates, although the yields and stereoselectivities were not very satisfactory. In addition, a tentative mechanism of the PPT-catalyzed aldol reaction of aldehyde with cyclohexanone was hypothesized (Scheme 4).41 Trypsin is one of the important serine proteases with its active center having a catalytic triad composed of His57, Asp102, and Ser195. Initially, the substrate cyclohexanone is stabilized through hydrogen bonding by the Asp–His dyad and the oxyanion hole which was formed by the backbone amide hydrogen atoms of Gly-193 and Ser-195 in the active site. Next, an enolate ion is formed after a proton on cyclohexanone is removed by the His residue. Then, a carbon–carbon bond is formed by the combination of the enolate ion with the aldehyde, which is protonated by the imidazolium cation. Eventually, the target aldol product is released from the oxyanion hole.
 | ||
| Scheme 4 Proposed mechanism for the trypsin-catalyzed aldol reaction. | ||
Furthermore, as one type of protease, AMP was also verified as a catalyst in the preparation of chiral aldol products through the transformations of aromatic aldehydes and cyclic or acyclic ketones by our group in 2013.19 AMP showed a remarkable substrate tolerance in line with the reactions of aromatic aldehydes with cyclic ketones covering 19 examples. Aromatic aldehyde substrates bearing various substituents and cyclic ketones with different sizes were successfully combined by the AMP to furnish the corresponding products in yields of up to 89% with dr of up to >99/1 and enantiomeric excess of up to 91% (Table 5, entry 5). Furthermore, in 2013, our group discovered that ficin from fig tree latex displayed promiscuous activity to catalyze the direct asymmetric aldol reactions of aromatic aldehydes and cyclic ketones although only two examples were exhibited (Table 5, entry 6).44
By comparing the catalytic effect of the different proteases listed above on direct asymmetric aldol reactions, it could be seen that AMP not only had powerful substrate adaptability, but also showed better catalytic activity and stereoselectivity than the other tested proteases in their promoted aldol reactions.
Anyway, some different types of proteases have been successfully introduced to promote the asymmetric aldol reactions of aromatic aldehydes with cyclic ketones. The vast majority of such experiments were performed in organic solvents in the presence of a small amount of water at ambient temperature. In addition, most of these proteases could tolerate a large scope of substrates and mainly offered anti-aldol products. Although some examples exhibited optimistic effects such as yields of up to 92% (Table 5, entry 1), dr of up to >99/1 (Table 5, entries 2 and 5), and ee of up to >99% (Table 5, entry 1), the general yields and enantioselectives were still relatively low. Another drawback was the requirement of long reaction times in these protease-catalyzed processes. In the further research, continued efforts are still needed to improve these disadvantages.
 | ||
| Scheme 5 The ficin-catalyzed direct asymmetric aldol reactions of aromatic aldehydes with heterocyclic ketones. | ||

2.3. The nucleases p1-catalyzed direct asymmetric aldol reactions
Nuclease p1 from Penicillium citrinum, a member of zinc-dependent endonucleases consisting of 270 amino acid residues with two disulfide bonds, cleaves single-stranded RNA and DNA into 5-mononucleotide.48–50 In 2011, our group found that nuclease p1 was a good catalyst for the direct asymmetric aldol reactions of aromatic aldehydes and cyclic ketones under organic, solvent-free conditions.51 Some control experiments were performed, which indicated that nuclease p1 was responsible for the direct asymmetric aldol reaction investigated. Some factors, such as the reaction medium, molar ratio of substrates, enzyme loading, water concentration and temperature, had impacts on the nuclease p1-catalyzed aldol reaction. The enantioselectivities of up to 99% ee and diastereoselectivities of up to >99/1 (anti/syn) were achieved in spite of the relatively low yields under the optimized conditions (Scheme 6). | ||
| Scheme 6 The nuclease p1-catalyzed aldol reactions of aromatic aldehydes and cyclic ketones. | ||
As already described in the previous section, in 2014, Lin and co-workers found that the asymmetric aldol addition of isatin and cyclohexanone could be catalyzed by some proteases. In the same study,47 they also reported that nuclease p1 from Penicillium citrinum could catalyze this type of aldol transformation. According to control experiments, no product was observed for the reaction with nuclease p1 denatured by ethylenediaminetetraacetic acid disodium salt dihydrate (EDTA) or in the absence of the enzyme. Finally, the best results (yields of up to 95%, dr of up to >99/1, ee of up to 82%) were obtained under organic solvent-free conditions after various isatin derivatives and cyclic ketones were investigated (Scheme 7).
 | ||
| Scheme 7 The nuclease p1-catalyzed asymmetric direct aldol reactions of isatin derivatives and cyclic ketones. | ||
2.4. Some miscellaneous aspects of the hydrolase-catalyzed direct asymmetric aldol reactions
In addition to lipase, protease and nuclease, some other members of hydrolase were also characterized with catalytic promiscuity in the direct asymmetric aldol reactions. Actually, D-aminoacylase was already discovered for the direct asymmetric aldol reaction of 4-nitrobenzaldehyde and acetone, in Wang's report in 2010.27 In this reaction, the yield of 35% was obtained; however, only 7% ee was detected. In 2014, L. Wang and coworkers found that a thermophilic esterase (APE1547) from the archaeon Aeropyrum pernix K1, a recombinant hyperthermophilic esterase, had catalytic ability in the asymmetric aldol reaction of 2-butanone and 4-nitrobenzaldehyde in organic solvents.29 Several influencing factors, including solvent, temperature, water content and substrate concentration, were optimized. Particularly, they found that n-heptane was superior to other polar solvents, which could be attributed to the unique enzyme structure of thermophilic esterase APE1547. This enzyme has a β-propeller domain, which is connected to the catalytic domain via two polypeptides. Hydrogen bonds and salt bridges with additional stability provided by hydrophobic forces can keep the two domains stable. Under the optimum conditions, the highest enzyme activity (38.1 μmol g−1 h−1) was obtained with 71.2% ee of the optically active secondary alcohol. A yield of 68.7% could be obtained after approximately 120 h according to the time course (Scheme 8).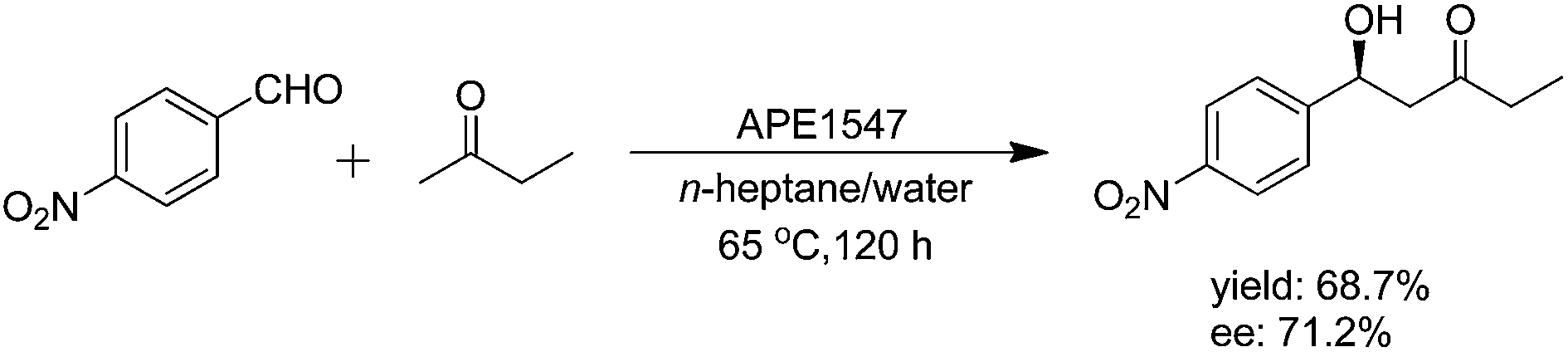 | ||
| Scheme 8 APE1547-catalyzed asymmetric aldol reaction of 2-butanone and 4-nitrobenzaldehyde. | ||
2.5. Brief summary of the hydrolase-catalyzed direct asymmetric aldol reactions
A series of hydrolases have been applied in the direct asymmetric aldol reactions. In particular, proteases and lipases were highlighted with prominent catalytic promiscuity. Moreover, some other hydrolases, such as nuclease p1, D-aminoacylase and esterase APE1547, were also reflected to own catalytic ability. According to these various direct asymmetric aldol reactions catalyzed by hydrolases in term of yields and stereoselectivities, the reactions between aromatic aldehydes and cyclic ketones appeared with better results, mainly affording anti-products. However, the results of aromatic aldehydes reacting with acyclic ketones were not satisfactory, which was probably due to the fact that short chain ketone molecules could not provide enough steric hindrance to give rise to stereoselectivity. Interestingly, some new cases already began emerging in succession such as the reactions of aromatic aldehydes with heterocyclic ketones and isatin derivatives with cyclic ketones. These cases of the enzymatic process afforded diverse chiral aldol products and extended the application of promiscuous hydrolases.It is worth mentioning that some catalytic mechanisms were proposed for these hydrolase-catalyzed direct asymmetric aldol reactions. Most of the mechanisms are similar which might be due to the fact that many hydrolases have highly similar structure of active site.25,52,53 For example, according to the mechanisms presented in the previous sections, the catalytic active site of each hydrolase includes a histidine, which plays the same role of capturing a proton from a ketone to allow it form an enolate. All of the proposed catalytic mechanisms of hydrolase-catalyzed asymmetric aldol reactions in this review include the following process: one substrate ketone is activated as the enolate anion by the residues on the active site; the enolate anion combines with the other substrate aldehyde, which is protonated by the imidazolium cation, and then the aldol product is released from the active site. Actually, the study of mechanism is important to clearly understand the promiscuous enzymatic process in essence. However, to date, all of the mechanisms of the hydrolase-catalyzed asymmetric aldol reactions are hypothesized, and more exact mechanisms need to be further verified by related experiments such as enzyme mutation and molecular docking.
3. The hydrolase-catalyzed asymmetric Michael additions
Michael addition, a typical case of conjugate additions, is one of the most fundamental and useful methods to produce new carbon–carbon bonds in organic synthesis via the 1,4-addition of nucleophiles to α,β-unsaturated carbonyl compounds.54–56 Employing the catalytic promiscuity of enzymes in Michael additions has aroused great interest for a long time in organic synthesis. As early as in the 1980s, Kitazume et al. had already noted that hydrolytic enzymes had the ability to catalyze the Michael additions of thiols and amines to triflourinate α,β-unsaturated carbonyl compounds in buffer solution.57,58 However, only in the last decade, more reports about enzymatic Michael additions began to emerge.55,59–69 The catalytic promiscuity of some hydrolases in the carbon–carbon bond formation by Michael additions has been attentively studied and has achieved delightful results. Nevertheless, hydrolase-catalyzed Michael additions in an asymmetric manner are, to date, few.In 2010, our group reported the first hydrolase-catalyzed asymmetric carbon–carbon Michael additions of a wide range of 1,3-dicarbonyl compounds and cyclohexanone to aromatic and heteroaromatic nitroolefins and cyclohexenone in organic media in the presence of water.70 Delightfully, the catalytic promiscuity of some lipases was reflected in this type of Michael addition (Table 7, entries 1–7). In consideration of yield and enantioselectivity, lipozyme TLIM (immobilized lipase from Thermomyces lanuginosus), a recyclable enzyme, was identified to be the most effective catalyst (Table 7, entry 3). Substrate extension demonstrated that lipozyme TLIM could endure different aromatic and heteroaromatic nitroalkenes, cyclohexenone, as well as other α,β-unsaturated aldehydes and ketones as the acceptors to react with 1,3-dicarbonyl compounds or cyclohexanone in DMSO in the presence of water, and enantioselectivities of up to 83% ee and yields of up to 85% were achieved (Table 8).
|
||||
|---|---|---|---|---|
| Entry | Enzyme | Time (h) | Yield (%) | ee (%) (s) |
| 1 | Lipase from porcine pancreas (PPL) | 75 | 78 | 4 |
| 2 | Lipase from porcine pancreas type II (PPL II) | 75 | 50 | 5 |
| 3 | Lipozyme TLIM | 75 | 75 | 17 |
| 4 | Lipase from Candida cylindracea | 75 | 63 | 11 |
| 5 | Lipase AYS“amano” Candida rugosa | 90 | 22 | 5 |
| 6 | Lipase PS “amano” SD from Burkholderia cepacia | 90 | 13 | 7 |
| 7 | Amano lipase AK, from Pseudomonas fluorescens | 90 | 35 | 2 |




















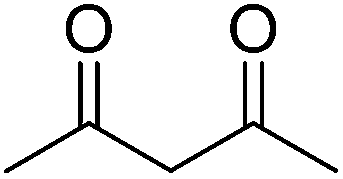


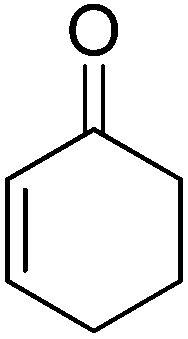
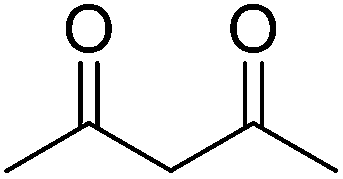
In 2012, our group surveyed another type of Michael addition, the reaction between 4-hydroxycoumarin and α,β-unsaturated enones, for the preparation of warfarin (an effective anticoagulant) and its derivatives by employing some lipases as catalysts (Table 9).71 Interestingly, lipase from porcine pancreas (PPL), one of the promiscuous hydrolases, was found to play multiple roles in organic reactions. PPL not only had the ability to promiscuously catalyze asymmetric aldol reactions, as previously presented in this study, but also showed better activity than other lipases in catalyzing the asymmetric Michael addition of 4-hydroxycoumarin and α,β-unsaturated enones. After systematically investigating the influence of the parameters on the synthesis of warfarin by the PPL-catalyzed Michael reaction, moderate to high yields (up to 95%) with certain enantioselectivities (up to 28% ee) were obtained in DMSO–water (9:1) at 25 °C (Table 9, entry 1). It was the first time a biocatalyst was used in the preparation of warfarin and its derivatives.
|
|||||
|---|---|---|---|---|---|
| Entry | Enzyme | Temp. (°C) | Yield (%) | ee (%) | Number of examples |
| 1 | Lipase from porcine pancreas (PPL) | 25–50 | 22–95 | Up to 28 | 9 |
| 2 | Amano lipase A from Aspergillus niger | 25 | 48 | 10 | 1 |
| 3 | Amano lipase M from Mucor javanicus | 25 | 42 | 6 | 1 |
| 4 | Lipase from Rhizopus niveus | 25 | 36 | 9 | 1 |
| 5 | Lipase from Candida cylindracea | 25 | 12 | 6 | 1 |

In comparison with the hydrolase-catalyzed direct asymmetric aldol reactions, the cases of the hydrolase-catalyzed asymmetric Michael additions are relatively few. Some lipases were adopted for the preparation of enantiomerically enriched products by Michael additions. For a few products, the yields of up to 95%, or ee of >99% were harvested, although modest yields with low enantioselectivities of most products were received.
4. The hydrolase-catalyzed direct asymmetric Mannich reactions
The Mannich reaction, one of the atom-economic and multicomponent coupling reactions involving an enolizable carbonyl compound and an imine in the reaction process, plays a very important role in the construction of carbon–carbon bonds.72–74 Although some enzyme-catalyzed Mannich reactions have been reported, the Mannich reactions catalyzed by an enzyme in an asymmetric manner are very limited.In 2012, our group reported the first hydrolase-catalyzed direct asymmetric three-component Mannich reaction by means of protease type XIV from Streptomyces griseus (SGP) in acetonitrile in the presence of water.75 Some control experiments indicated that the specific natural fold of SGP was necessary for its stereoselectivity in the Mannich reaction. For example, the reaction with urea-denatured SGP as a catalyst gave the product in a high yield of 87% with only 8% ee. Moreover, 250 mM Cu2+ or Ag+ almost completely destroyed the selectivity of SGP in the Mannich reaction. Some parameters were attentively optimized by exploring the influence of solvent, water content, pH, molar ratio of the substrates and temperature. Many electrophilic and nucleophilic species were available as substrates in the SGP-catalyzed Mannich reactions such as various substituted aromatic aldehydes, various substituted arylamines, heteroatom-containing cycloketones and linear chain ketones. Among these SGP-catalyzed Mannich reactions, the highest yield of up to 92%, the best enantioselectivity of up to 88% ee and diastereoselectivity of up to 92/8 were achieved (Table 10, entry 1).
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Enzyme | Medium | Temp. (°C) | Time (h) | Yield (%) | dr (syn/anti) | ee (%) | Number of examples | Ref. |
| a SGP: protease type XIV from Streptomyces griseus.b AMA: acylase I from Aspergillus melleus. | |||||||||
| 1 | SGPa | MeCN/water | 30 | 94–168 | 24–92 | 40/60–92/8 | 33–88 | 17 | 75 |
| 2 | AMAb | MeCN/buffer | 30 | 96–192 | 31–82 | 43/57–90/10 | 23–89 | 17 | 76 |

Very recently, our group reported another hydrolase-catalyzed asymmetric Mannich reaction employing acylase I (aminoacylase) from Aspergillus melleus (AMA) as a catalyst. Acylase is one of the top 10 enzymes used in biotechnology.76,77 The activity and stereoselectivity of the AMA in the Mannich reaction could be improved by adjusting solvent, pH, water content, temperature, molar ratio of the substrates and enzyme loading. Under the optimized conditions, the enantioselectivities of up to 89% ee, diastereoselectivities of up to 90/10 dr and yields of up to 82% were achieved (Table 10, entry 2).
5. The hydrolase-catalyzed direct asymmetric Morita–Baylis–Hillman reaction
The Morita–Baylis–Hillman reaction (MBH reaction), an emerging and fascinating carbon–carbon bond-forming reaction, can be identified as a carbon–carbon forming reaction between the α-position of activated conjugated carbonyl compounds and carbon electrophiles in the presence of a suitable nucleophilic catalyst.78–83 In 2007, Reetz and co-workers reported the first promiscuous enzyme-catalyzed asymmetric MBH transformation of cyclohexenone and p-nitrobenzaldehyde.84 Among the tested hydrolases, only hog pancreas lipase exhibited a certain enantioselectivity, giving the product in 2% ee with the conversion of 7% (Scheme 9). A relatively long reaction time of 5 days was required, and the results (enantioselectivity and activity) were not very satisfactory. However, this observation extended the types of hydrolase-catalyzed asymmetric reactions. | ||
| Scheme 9 MBH reaction of cyclohexenone and p-nitrobenzaldehyde catalyzed by hog pancreas lipase. | ||
6. Conclusions and prospects
In this review, we highlighted asymmetric carbon–carbon bond forming transformations catalyzed by promiscuous hydrolases. All of these hydrolases were commercially available (except thermophilic esterase (APE1547) from the archaeon Aeropyrum pernix K1, a recombinant hyperthermophilic esterase) and displayed their catalytic activity without co-factors. Most of the reactions could be realized in organic solvents in the presence of water as an optimum content of water is essential for the enzyme to keep its catalytic activity.85,86 Generally, acetonitrile exhibited outstanding performance among the organic solvents, but the reason for this phenomenon needs to be further verified. Moreover, the reaction conditions were mild. These hydrolase-catalyzed asymmetric reactions could be successfully conducted at around 30 °C, although a long reaction time was needed in many cases. Gratifyingly, some enantiomerically enriched products were obtained in excellent yields with the help of the catalytic promiscuity of hydrolases. However, in most cases, only low to moderate yields and low stereoselectivities were achieved. Interestingly, some hydrolases possess various catalytic promiscuities. For instance, PPL has been employed as a catalyst for both asymmetric aldol reactions and Michael additions. Nevertheless, to date, the types of hydrolase-catalyzed asymmetric carbon–carbon bond formations are only limited to aldol, Michael, Mannich and MBH reactions. Therefore more types of asymmetric reactions need to be explored with the catalytic promiscuity of hydrolases. In addition, it is meaningful that the catalytic mechanisms of some hydrolase-catalyzed aldol reactions were proposed, which provided a better understanding of the catalytic process. However, the research of the mechanism was insufficient, and none of the proposed mechanisms thus far have explained the observed stereoselectivity. Thus, further in-depth mechanism studies are necessary to contribute to a better understanding of the relationship between catalytic promiscuity and the specificity of enzymes.Although many drawbacks exist in this aspect of biocatalysis, which need to be further improved, hydrolase-catalyzed asymmetric carbon–carbon bond formation for the preparation of the enantiomerically enriched compounds presents a useful methodology in organic synthesis. Consequently, the new insight obtained from the study of catalytic promiscuity of hydrolases in asymmetric carbon–carbon bond formation is crucial not only for the provision of new chiral catalysts in organic synthesis, but also for new enzyme design by directed evolution techniques and protein engineering.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (no. 21276211 and no. 21472152).References
- M. S. Humble and P. Berglund, Eur. J. Org. Chem., 2011, 2011, 3391–3401 CrossRef CAS.
- E. Busto, V. Gotor-Fernandez and V. Gotor, Chem. Soc. Rev., 2010, 39, 4504–4523 RSC.
- K. Hult and P. Berglund, Trends Biotechnol., 2007, 25, 231–238 CrossRef CAS PubMed.
- A. Babtie, N. Tokuriki and F. Hollfelder, Curr. Opin. Chem. Biol., 2010, 14, 200–207 CrossRef CAS PubMed.
- S. D. Copley, Curr. Opin. Chem. Biol., 2003, 7, 265–272 CrossRef CAS.
- I. Nobeli, A. D. Favia and J. M. Thornton, Nat. Biotechnol., 2009, 27, 157–167 CrossRef CAS PubMed.
- O. Khersonsky and D. S. Tawfik, Annu. Rev. Biochem., 2010, 79, 471–505 CrossRef CAS PubMed.
- G. Seoane, Curr. Org. Chem., 2000, 4, 283–304 CrossRef CAS.
- V. Resch, J. H. Schrittwieser, E. Siirola and W. Kroutil, Curr. Opin. Biotechnol., 2011, 22, 793–799 CrossRef PubMed.
- M. Beigi, S. Waltzer, A. Fries, L. Eggeling, G. A. Sprenger and M. Müller, Org. Lett., 2013, 15, 452–455 CrossRef CAS PubMed.
- M. Müller, Adv. Synth. Catal., 2012, 354, 3161–3174 CrossRef.
- M. Brovetto, D. Gamenara, P. Saenz Méndez and G. A. Seoane, Chem. Rev., 2011, 111, 4346–4403 CrossRef CAS PubMed.
- Y. Ding, H. Huang and Y. Hu, Chin. J. Org. Chem., 2013, 33, 905–914 CrossRef CAS.
- J. I. G. Cadogan, A. A. Doyle, I. Gosney, P. K. G. Hodgson and P. Thorburn, Enantiomer, 1997, 2, 81–98 CAS.
- M. M. Heravi and V. Zadsirjan, Tetrahedron: Asymmetry, 2013, 24, 1149–1188 CrossRef CAS PubMed.
- D. Gryko and D. Walaszek, in Stereoselective Organocatalysis, John Wiley & Sons, Inc., 2013, pp. 81–127, DOI:10.1002/9781118604755.ch03.
- B. Voigt, U. Scheffler and R. Mahrwald, Chem. Commun., 2012, 48, 5304–5306 RSC.
- M. Markert and R. Mahrwald, Chem.–Eur. J., 2008, 14, 40–48 CrossRef CAS PubMed.
- Y. Yuan, Z. Guan and Y.-H. He, Sci. China: Chem., 2013, 56, 939–944 CrossRef CAS.
- R. Mestres, Green Chem., 2004, 6, 583–603 RSC.
- C. Branneby, P. Carlqvist, A. Magnusson, K. Hult, T. Brinck and P. Berglund, J. Am. Chem. Soc., 2002, 125, 874–875 CrossRef PubMed.
- X. Ge, Y.-F. Lai and X.-Z. Chen, Youji Huaxue, 2013, 33, 1686–1696 CrossRef CAS.
- F. Hasan, A. A. Shah and A. Hameed, Enzyme Microb. Technol., 2006, 39, 235–251 CrossRef CAS PubMed.
- R. C. Tang, Z. Guan, Y. H. He and W. Zhu, J. Mol. Catal. B: Enzym., 2010, 63, 62 CrossRef CAS PubMed.
- B.-K. Liu, Q. Wu and X.-F. Lin, Curr. Org. Chem., 2010, 14, 1966–1988 CrossRef.
- C. Li, X.-W. Feng, N. Wang, Y.-J. Zhou and X.-Q. Yu, Green Chem., 2008, 10, 616–618 RSC.
- C. Li, Y.-J. Zhou, N. Wang, X.-W. Feng, K. Li and X.-Q. Yu, J. Biotechnol., 2010, 150, 539–545 CrossRef CAS PubMed.
- Z.-B. Xie, N. Wang, L.-H. Zhou, F. Wan, T. He, Z.-G. Le and X.-Q. Yu, ChemCatChem, 2013, 5, 1935–1940 CrossRef CAS.
- H.-R Wang, Z. Wang, H. Zhang, G. Chen and L. Wang, Green Chem. Lett. Rev., 2014, 7, 145–149 CrossRef.
- Z.-B. Xie, N. Wang, G.-F. Jiang and X.-Q. Yu, Tetrahedron Lett., 2013, 54, 945–948 CrossRef CAS PubMed.
- B.-H. Xie, J. Zheng, Y.-L. Chen, J.-F. Cao, Y. Yang, Z. Guan and Y.-H. He, Z. Naturforsch., 2014, 69c, 170–180 Search PubMed.
- T. Sengoku, T. Takemura, E. Fukasawa, I. Hayakawa and H. Kigoshi, Tetrahedron Lett., 2009, 50, 325–328 CrossRef CAS PubMed.
- M. S. Frasinyuk, S. P. Bondarenko, V. P. Khilya, C. Liu, D. S. Watt and V. M. Sviripa, Org. Biomol. Chem., 2015, 13, 1053–1067 CAS.
- M. A. Ciufolini, Can. J. Chem., 2014, 92, 186–193 CrossRef CAS.
- Z. Guan, J.-P. Fu and Y.-H. He, Tetrahedron Lett., 2012, 53, 4959–4961 CrossRef CAS PubMed.
- F. Bordusa, Chem. Rev., 2002, 102, 4817–4868 CrossRef CAS PubMed.
- A. Zaks and A. Klibanov, Science, 1984, 224, 1249–1251 CAS.
- A.-X. Yan, G.-L. Tian and Y.-H. Ye, Prog. Chem., 2001, 13, 203–208 CAS.
- M. López-Iglesias, E. Busto, V. Gotor and V. Gotor-Fernández, Adv. Synth. Catal., 2011, 353, 2345–2353 CrossRef.
- Y.-H. He, H.-H. Li, Y.-L. Chen, Y. Xue, Y. Yuan and Z. Guan, Adv. Synth. Catal., 2012, 354, 712–719 CrossRef CAS.
- Y.-L. Chen, W. Li, Y. Liu, Z. Guan and Y.-H. He, J. Mol. Catal. B: Enzym., 2013, 87, 83–87 CrossRef CAS PubMed.
- H.-H. Li, Y.-H. He and Z. Guan, Catal. Commun., 2011, 12, 580–582 CrossRef CAS PubMed.
- B.-H. Xie, W. Li, Y. Liu, H.-H. Li, Z. Guan and Y.-H. He, Tetrahedron, 2012, 68, 3160–3164 CrossRef CAS PubMed.
- J.-P. Fu, N. Gao, Y. Yang, Z. Guan and Y.-H. He, J. Mol. Catal. B: Enzym., 2013, 97, 1–4 CrossRef CAS PubMed.
- W. C. Sumpter, Chem. Rev., 1944, 34, 393–434 CrossRef CAS.
- Y.-C. Liu, R. Zhang, Q.-Y. Wu, Q. Chen and G.-F. Yang, Org. Prep. Proced. Int., 2014, 46, 317–362 CrossRef CAS.
- Z.-Q. Liu, Z.-W. Xiang, Z. Shen, Q. Wu and X.-F. Lin, Biochimie, 2014, 101, 156–160 CrossRef CAS PubMed.
- A. Volbeda, A. Lahm, F. Sakiyama and D. Suck, EMBO J., 1991, 10, 1607–1618 CAS.
- A. Lahm, A. Volbeda and D. Suck, J. Mol. Biol., 1990, 215, 207–210 CrossRef CAS.
- W. N. Lipscomb and N. Sträter, Chem. Rev., 1996, 96, 2375–2434 CrossRef CAS PubMed.
- H.-H. Li, Y.-H. He, Y. Yuan and Z. Guan, Green Chem., 2011, 13, 185–189 RSC.
- P. Carter and J. A. Wells, Nature, 1988, 332, 564–568 CrossRef CAS PubMed.
- C. Oinonen and J. Rouvinen, Protein Sci., 2000, 9, 2329–2337 CrossRef CAS PubMed.
- A. Michael, J. Appl. Chem., 1887, 35, 349–356 Search PubMed.
- M. Svedendahl, K. Hult and P. Berglund, J. Am. Chem. Soc., 2005, 127, 17988–17989 CrossRef CAS PubMed.
- M. Svedendahl, B. Jovanović, L. Fransson and P. Berglund, ChemCatChem, 2009, 1, 252–258 CrossRef CAS.
- T. Kitazume, T. Ikeya and K. Murata, J. Chem. Soc., Chem. Commun., 1986, 17, 1331–1333 RSC.
- T. Kitazume, K. Murata, Y. Kokusho and S. Iwasaki, J. Fluorine Chem., 1988, 39, 75–86 CrossRef CAS.
- O. Torre, I. Alfonso and V. Gotor, Chem. Commun., 2004, 15, 1724–1725 RSC.
- Y. Cai, X.-F. Sun, N. Wang and X.-F. Lin, Synthesis, 2004, 5, 671–674 Search PubMed.
- V. Gotor-Fernández, E. Busto and V. Gotor, Adv. Synth. Catal., 2006, 348, 797–812 CrossRef.
- J.-M. Xu, F. Zhang, B.-K. Liu, Q. Wu and X.-F. Lin, Chem. Commun., 2007, 20, 2078–2080 RSC.
- G. A. Strohmeier, T. Sović, G. Steinkellner, F. S. Hartner, A. Andryushkova, T. Purkarthofer, A. Glieder, K. Gruber and H. Griengl, Tetrahedron, 2009, 65, 5663–5668 CrossRef CAS PubMed.
- P. Carlqvist, M. Svedendahl, C. Branneby, K. Hult, T. Brinck and P. Berglund, ChemBioChem, 2005, 6, 331–336 CrossRef CAS PubMed.
- Y.-F. Lai and P.-F. Zhang, Res. Chem. Intermed., 2013, 1–6, DOI:10.1007/s11164-013-1512-6.
- E. Zandvoort, E. M. Geertsema, B.-J. Baas, W. J. Quax and G. J. Poelarends, Angew. Chem., Int. Ed., 2012, 51, 1240–1243 CrossRef CAS PubMed.
- Y. Miao, E. M. Geertsema, P. G. Tepper, E. Zandvoort and G. J. Poelarends, ChemBioChem, 2013, 14, 191–194 CrossRef CAS PubMed.
- T. Narancic, J. Radivojevic, P. Jovanovic, D. Francuski, M. Bigovic, V. Maslak, V. Savic, B. Vasiljevic, K. E. O'Connor and J. Nikodinovic-Runic, Bioresour. Technol., 2013, 142, 462–468 CrossRef CAS PubMed.
- E. M. Geertsema, Y. Miao, P. G. Tepper, P. de Haan, E. Zandvoort and G. J. Poelarends, Chem.–Eur. J., 2013, 19, 14407–14410 CrossRef CAS PubMed.
- J.-F. Cai, Z. Guan and Y.-H. He, J. Mol. Catal. B: Enzym., 2011, 68, 240–244 CrossRef CAS PubMed.
- B.-H. Xie, Z. Guan and Y.-H. He, J. Chem. Technol. Biotechnol., 2012, 87, 1709–1714 CrossRef CAS.
- W. Mario, Prog. Chem. Org. Nat. Prod., 2012, 96, 1–197 Search PubMed.
- L.-W. Xu, C.-G. Xia and L. Li, J. Org. Chem., 2004, 69, 8482–8484 CrossRef CAS PubMed.
- K. Nagata, Y. Kuga, A. Higashi, A. Kinoshita, T. Kanemitsu, M. Miyazaki and T. Itoh, J. Org. Chem., 2013, 78, 7131–7136 CrossRef CAS PubMed.
- Y. Xue, L.-P. Li, Y.-H. He and Z. Guan, Sci. Rep., 2012, 2, 761, DOI:10.1038/srep00761.
- Z. Guan, J. Song, Y. Xue, D.-C. Yang and Y.-H. He, J. Mol. Catal. B: Enzym., 2015, 111, 16–20 CrossRef CAS PubMed.
- Y. Tewari, Appl. Biochem. Biotechnol., 1990, 23, 187–203 CrossRef CAS.
- K. M. ortia, Z. Suzuki and H. Hirose, Bull. Chem. Soc. Jpn., 1968, 41, 2815 CrossRef.
- A. B. Baylis and M. E. D. Hillman, German patent 2155113, 1972.
- D. Basavaiah, P. Dharma Rao and R. Suguna Hyma, Tetrahedron, 1996, 52, 8001–8062 CrossRef CAS.
- D. Basavaiah, A. J. Rao and T. Satyanarayana, Chem. Rev., 2003, 103, 811–892 CrossRef CAS PubMed.
- V. K. Aggarwal, S. Y. Fulford and G. C. Lloyd-Jones, Angew. Chem., Int. Ed., 2005, 44, 1706–1708 CrossRef CAS PubMed.
- S. E. Drewes and G. H. P. Roos, Tetrahedron, 1988, 44, 4653–4670 CrossRef CAS.
- M. T. Reetz, R. Mondière and J. D. Carballeira, Tetrahedron Lett., 2007, 48, 1679–1681 CrossRef CAS PubMed.
- A. Zaks and A. M. Klibanov, J. Biol. Chem., 1988, 263, 3194–3201 CAS.
- Y. N. Sardessai and S. Bhosle, Biotechnol. Prog., 2004, 20, 655–660 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |