
Organocatalytic asymmetric Michael addition of aldehydes and ketones to nitroalkenes catalyzed by adamantoyl l-prolinamide†
Abstract
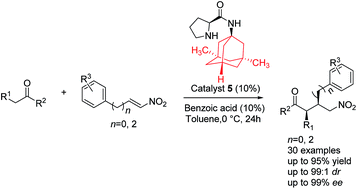
A series of adamantoyl L-prolinamides have been synthesized. These compounds have been found to be highly efficient organocatalysts for the Michael addition of aldehydes and ketones to nitroalkenes. Under the optimized reaction conditions, the corresponding Michael adducts were obtained in good yields (up to 95%), excellent enantioselectivities (up to 99% ee) and moderate diastereoselectivities.
 Please wait while we load your content...
Please wait while we load your content...

