
Efficient synthesis of zirconium poly(styrene-phenylvinylphosphonate)phosphate-supported proline as a recyclable catalyst for direct asymmetric aldol reactions in water†
Chuanlong Wu*a,
Xiaoqin Longa,
Xiangkai Fub,
Guangwei Wang*c and
Zakaria. A. Mirzac
aChongqing Unis Chemical Co, Ltd, National Enterprise Technology Center, Chongqing, 402161, P. R. China. E-mail: wcl110@swu.edu.cn
bCollege of Chemistry and Chemical Engineering Southwest University, Chongqing, 400715, P. R. China
cChongqing Institute of Green and Intelligent Technology (CIGIT), Chinese Academy of Sciences, Chongqing, 400714, P. R. China. Fax: +86 2368254000; Tel: +86 2368253704
First published on 4th December 2014
Abstract
A completely non-chromatographic and highly large-scale adaptable synthesis of zirconium poly(styrene-phenylvinylphosphonate)phosphate-supported L-proline (ZrPS-PVPA-Pr) has been developed in only three steps overall. Catalyst 1c was determined to be efficient for the asymmetric direct aldol reaction (only 0.5 mol% of 1c was used) in the presence of water at room temperature, with an enantiomeric excess as high as 99%. In addition, water was identified as one of the most significant reaction condition parameters due to the characteristics of the organic–inorganic hybrid catalyst support. Catalyst 1c was easily recovered by simple filtration and could be reused at least six times with little loss of activity and enantioselectivity. Catalyst 1c can be used efficiently on a large-scale while maintaining the enantioselectivities of the aldol reactions. Therefore, this method has the potential for application in industry.
Introduction
The increasing quantities of industrial waste exert a significant and serious impact on the environment and force the modern chemical industry toward using clean processes due to the increasing demand of chemicals in various applications.1–3 Therefore, to resolve this problem, the development and application of new types of heterogeneous catalysts for organic reactions is an important approach.Industrially, condensation reactions are of great importance in the production of a number of key compounds. The direct asymmetric aldol reaction is one of the most important carbon–carbon bond-forming reactions and has been widely used in constructing natural and non-natural products.4,5 Since the early reports in the 1970s on the L-proline catalysed intramolecular aldol reactions6,7 and the discovery by List et al. that L-proline can mimic type I aldolase to enantioselectively catalyse intermolecular aldol reactions,8 interest in organocatalysis has significantly increased in the past few years due to the novelty of the concept and unique activation modes.9–19 Many homogeneous chiral organocatalysts have been prepared, and many of these catalysts are known to be highly effective in asymmetric aldol reactions.20,21 However, few examples of these catalysts have been developed for use in industrial processes, which is due to the un-avoidable drawbacks of homogeneous catalytic processes (e.g., short lifetime, lower structural and thermal stability, and difficulties in separation and recovery of highly expensive chiral catalysts), as well as concerns regarding product purity. Therefore, recycling and reuse of these expensive catalyst systems become important aspects. In addition, the separation of catalysts from product streams poses economic and environmental challenges.22,23 Product separation, catalyst recovery, and resistance to drastic operational conditions, large-scale reactions are advantageous features of heterogeneous catalysts. A combination of these features would be desirable for establishing an ideal catalytic system.24
Currently, the immobilization of a chiral organocatalyst has been widely investigated on inorganic or organic supports, including mesoporous materials,25 metals,26 layered compounds,27 polymers28 and dendrimers.29 However, the development of practical solid support-immobilized asymmetric catalysts has proven highly challenging due to lower enantioselectivities or efficiencies and number of use cycles. Therefore, new types of heterogeneous catalyst systems need to be developed.
In recent years, the zirconium phosphates and zirconium phosphonate field has been actively investigated, and the focus has primarily been on adsorbents,30 inorganic ion exchangers,31 intercalation chemistry,32 catalyst supports and catalysts.33,34 The stable amorphous L-proline-functionalized zirconium methyl-and/or phenylphosphonates were prepared to catalyse the direct asymmetric aldol addition with high yield, diastereoselectivity and enantiomeric excess.35,36 The zirconium phosphate–phosphonate hybrid materials also have been extensively explored due to the large surface area, high thermal stability, good acid and chemical resistance, and advantage of that all of the organic groups were located on the surface of layers, interlamellar region and interlayer surface despite crystalline, semi-crystalline or amorphous nature of the solid.37
According to the theory and technique of hybridization or composition material, a hybrid or composite material exhibits new properties along with the original characteristics of each component. For the purpose of trying to synthesize a new type of organic–inorganic hybrid catalyst support that possesses quite different properties from either common organic support such as polystyrene or pure inorganic supports as silica gel, alumina, molecule sieve and zirconium phosphates or phosphonates, the frameworks of organic–inorganic hybrids ZrPS-PVPA-Pr were easily prepared in three steps overall. In addition, the method where the chiral organocatalyst is anchored onto ZrPS-PVPA (organic–inorganic hybrid support) has been rarely reported (Fig. 1).
 | ||
| Fig. 1 Ideal structural model of ZrPS-PVPA-Pr. | ||
In line with the “green chemistry”, effective and environmentally benign synthetic methodology is often regarded as a goal in modern organic chemistry. The use of water, which has some advantages (i.e., cost, safety, and environmental impact), as a reaction solvent rather than an organic solvent is preferred to decrease environmental contamination in addition to the use of a catalyst support.38–40 In addition, the surface of the supports as well as the caves, holes, pores, micropores and, channels in the supports may provide microenvironmental effects that exhibit different catalytic performances for asymmetric aldol reactions compared to those using either pure polystyrene or inorganic supports. In this paper, we report a new type of heterogeneous aldol reaction catalyst system using ZrPS-PVPA as a support, and this reaction proceeded efficiently in the presence of water at room temperature (Scheme 1).
 | ||
| Scheme 1 Synthesis of catalysts 1c and 1d. | ||
Experimental
Materials and instrumentation
Hydroxyproline, styrene, styrene phosphoric acid and, cinnamic acid, were supplied by Alfa Aesar. All of the chemicals were used as received unless otherwise noted. The reagent grade solvents were distilled prior to use. The reactions were monitored by thin-layer chromatography (TLC) carried out on 0.25 mm silica gel plates, which were visualized using UV light and/or by staining with ethanolic phosphomolybdic acid (PMA) and/or ninhydrin in an ethanol stain.Flash column chromatography was performed on silica gel (200–300 mesh). FT-IR spectra were recorded using KBr pellets on a Bruker RFS100/S spectrophotometer (USA) in the range of 4000–400 cm−1. The NMR spectra were recorded on a 300 MHz instrument. Chemical shifts (δ) are provided in ppm relative to TMS as the internal reference, and coupling constants (J) are reported in Hz. TG analysis was performed on a SBTQ600 Thermal Analyzer (USA) with a heating rate of 20 °C min−1 from 25 to 1000 °C under flowing N2 (100 mL min−1). SEM was performed on a KYKY-EM3 200 (KYKY, China) microscope, with a resolution of 6.0 nm (30 kV) and a magnification range of 15× to 250![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000×. TEM images were obtained on a TECNAI10 (PHILIPS, Holland) apparatus, with a line resolution of 0.144 nm, and a point resolution of 0.282 nm, and magnification range of 25× to 700000×. The BET surface areas were determined using N2 sorption data measured at 77 K (Quantachrome Autosorb-1). The sample was degassed at 100 °C for 8 h prior to obtaining the measurements. The pore size distribution curves were obtained from a desorption isotherm using the BJH method. Melting points were measured on a digital melting-point apparatus. Mass spectra (MS) were measured on HCT Bruker ESQUIRE LC/MS spectrometer with electrospray ionization. The elemental nitrogen content of the catalyst was determined using a Vario EL element analyser (Germany), with an Ar carrier gas. Analytical high performance liquid chromatography (HPLC) was carried out on an Agilent 1200 instrument using Chiralpak AD (4.6 mm × 250 mm) or Chiralcel OD-H (4.6 mm × 250 mm) columns. Optical rotations were measured on a JASCO P-1010 Polarimeter at λ = 589 nm.
000×. TEM images were obtained on a TECNAI10 (PHILIPS, Holland) apparatus, with a line resolution of 0.144 nm, and a point resolution of 0.282 nm, and magnification range of 25× to 700000×. The BET surface areas were determined using N2 sorption data measured at 77 K (Quantachrome Autosorb-1). The sample was degassed at 100 °C for 8 h prior to obtaining the measurements. The pore size distribution curves were obtained from a desorption isotherm using the BJH method. Melting points were measured on a digital melting-point apparatus. Mass spectra (MS) were measured on HCT Bruker ESQUIRE LC/MS spectrometer with electrospray ionization. The elemental nitrogen content of the catalyst was determined using a Vario EL element analyser (Germany), with an Ar carrier gas. Analytical high performance liquid chromatography (HPLC) was carried out on an Agilent 1200 instrument using Chiralpak AD (4.6 mm × 250 mm) or Chiralcel OD-H (4.6 mm × 250 mm) columns. Optical rotations were measured on a JASCO P-1010 Polarimeter at λ = 589 nm.
Catalyst synthesis
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O). (The mass fractions of the C, H and N, elements were determined by elemental analysis to be 39.88 mmol g−1, 47.50 mmol g−1 and 0.125 mmol g−1, respectively.)O). (The mass fractions of the C, H and N, elements were determined by elemental analysis to be 55.20 mmol g−1, 61.09 mmol g−1, and 0.182 mmol g−1, respectively.)
O). (The mass fractions of the C, H and N, elements were determined by elemental analysis to be 39.88 mmol g−1, 47.50 mmol g−1 and 0.125 mmol g−1, respectively.)O). (The mass fractions of the C, H and N, elements were determined by elemental analysis to be 55.20 mmol g−1, 61.09 mmol g−1, and 0.182 mmol g−1, respectively.)General procedure for the direct aldol reaction using catalyst 1c in the presence of water
To a mixture of catalyst 1c (10 mg, 0.125 mmol g−1 hydroxy-L-proline, determined according to N content of elemental analysis), cyclohexanone (1 mmol) and water (1 mL) were, stirred for 10 min at room temperature, and then aldehyde (0.25 mmol) was added. The reaction was monitored by TLC, and the mixture was filtered under vacuum and dried over anhydrous MgSO4. Diastereoselectivity was determined by 1HNMR or HPLC analysis of the crude aldol products. The enantiomeric excess (ee) was determined by chiral-phase HPLC analysis of the pure anti-product which was purified by flash column chromatography (petroleum ether:EtOAC = 2:1). The absolute configuration of the aldol products was extrapolated by comparison of the HPLC-data with known literature data.
Results and discussion
Characterisation of the catalyst
| Sample | IR, ν/cm−1 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1c | 3405 | 3082 | 3028 | 2924 | 1686 | 1493 | 1244 | 1025 | 756 | ||
| 1d | 3405 | 3059 | 3025 | 2923 | 1686 | 1453 | 1247 | 1029 | 1025 | 999 | 698 |
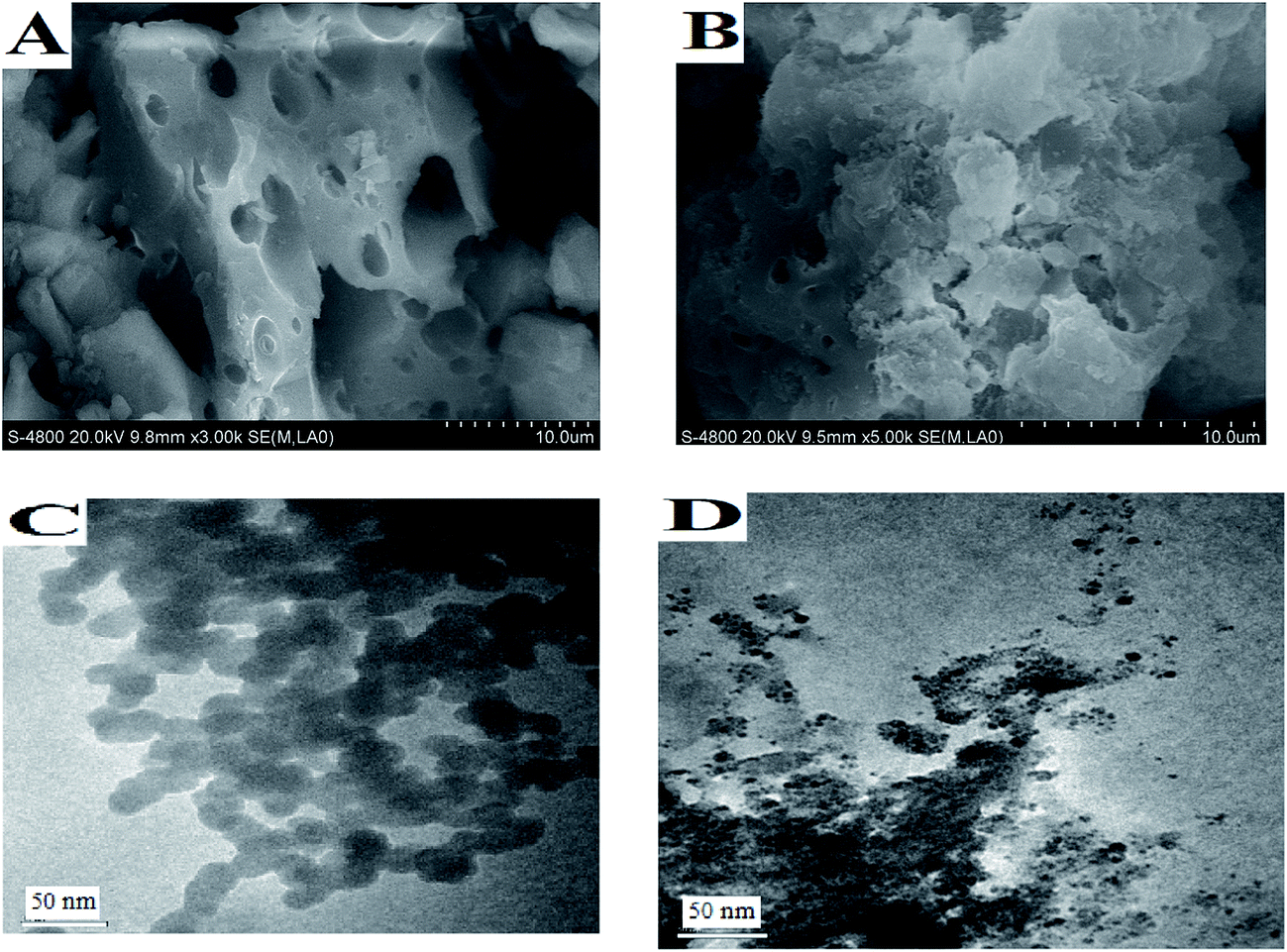 | ||
| Fig. 2 SEM (A: 1c; B: 1d) and TEM (C: 1c; D: 1d) analysis of catalysts 1c and 1d. | ||
Using the X-ray diffractogram measurements of compounds 1c and 1d (Fig. 3), the layer spacing was calculated. 1c exhibited a larger layer spacing of 30.026 nm compared to pure organic zirconium phosphate 1d (22.071 nm), which is due to changes in the electronic structure of the catalyst supports by addition of inorganic zirconium phosphate. The results indicated that the catalyst and substrate interact sufficiently in the adequate microenvironment among the layers of the heterogeneous catalyst bulk, as shown in the SEM and TEM images in Fig. 2.
 | ||
| Fig. 3 XRD pattern of catalysts 1c and 1d. | ||
| Catalyst | Times | C (%) | H (%) | N (%) | Surface area (cm2 g−1) | Pore volume (×10−3 cm3 g−1) | Average pore diameter (nm) |
|---|---|---|---|---|---|---|---|
| 1c | 1 | 47.80 | 4.685 | 0.176 | 23.08 | 9.719 | 8.422 |
| 2 | 47.91 | 4.824 | 0.174 | ||||
| 1d | 1 | 66.28 | 6.032 | 0.25 | 4.50 | 2.096 | 9.313 |
| 2 | 66.20 | 6.185 | 0.26 |
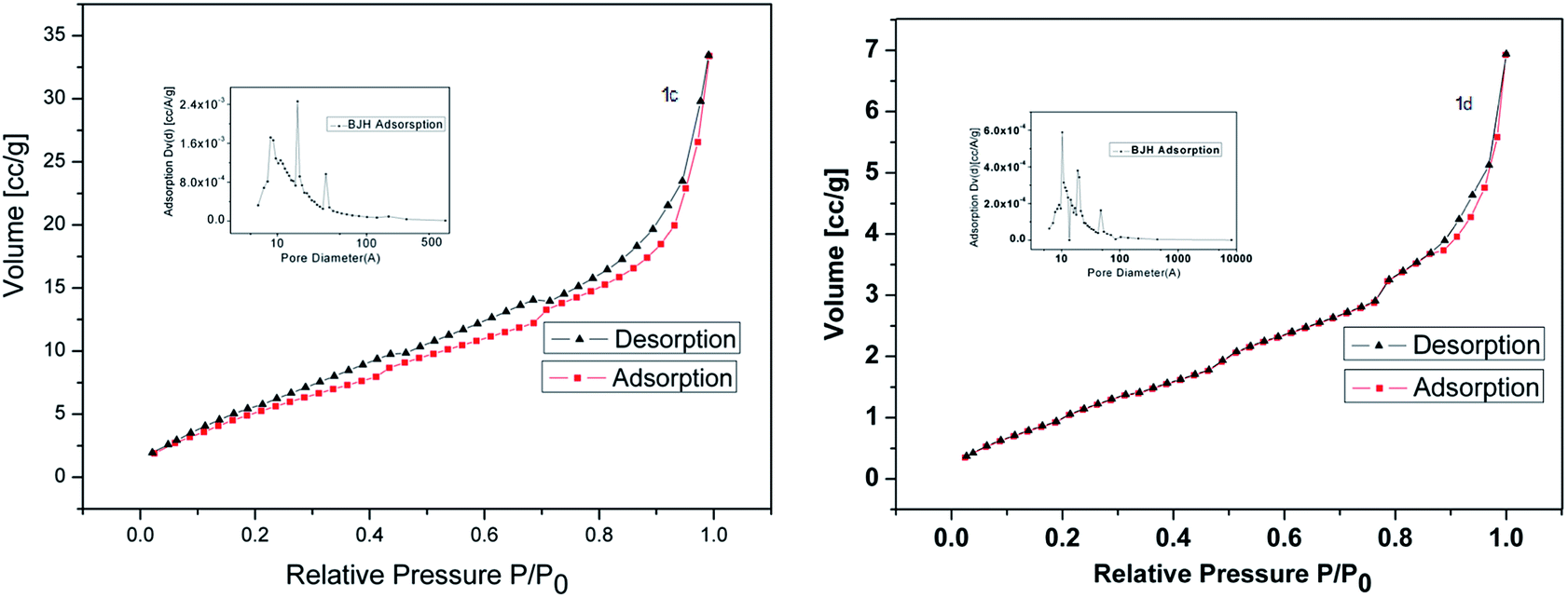 | ||
| Fig. 4 N2 adsorption–desorption analysis of catalysts 1c and 1d. | ||
 | ||
| Fig. 5 TG curves of catalyst 1c. | ||
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | Cat. loading [mg] | Solvents | Yieldb (%) | anti:sync |
eed (%) |
| a The reaction was performed with p-nitrobenzaldehyde (0.25 mmol), cyclohexanone (1.0 mmol), catalyst 1c and 1d (100 mg 0.125 mmol g−1), in the solvent (1.0 mL) at room temperature.b Isolated yield after chromatography on silica gel.c Determined by chiral 1HNMR analysis, major product is anti.d Determined by chiral HPLC analysis of the anti-product. | ||||||
| 1 | 1c | 100 | — | 60 | 53:47 |
44 |
| 2 | 1c | 100 | THF | 55 | 54:46 |
42 |
| 3 | 1c | 100 | CHCl3 | 53 | 63:37 |
36 |
| 4 | 1c | 100 | DMSO | 65 | 68:32 |
43 |
| 5 | 1c | 100 | n-C6H14 | 45 | 76:24 |
65 |
| 6 | 1c | 100 | Water | 99 | 85:15 |
93 |
| 7 | 1d | 100 | — | 58 | 56:44 |
14 |
| 8 | 1d | 100 | THF | 55 | 54:46 |
37 |
| 9 | 1d | 100 | CHCl3 | 52 | 50:50 |
— |
| 10 | 1d | 100 | DMSO | 89 | 84:14 |
93 |
| 11 | 1d | 100 | n-C6H14 | 50 | 61:39 |
30 |
| 12 | 1d | 100 | Water | 85 | 69:31 |
83 |

Then, we investigated the effects of different amounts of catalyst 1c on the reaction of cyclohexanone with p-nitrobenzaldehyde (Table 4). By using 10 mg of catalyst 1c (0.5 mol%, 0.125 mmol g−1), we obtained a good yield with excellent stereoselectivity (Table 4, entry 7). Therefore, the optimized amount of catalyst 1c loading was chosen to be 10 mg (0.5 mol%, 0.125 mmol g−1).
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | Cat loading [mg] | Solvents | Yieldb (%) | anti:sync |
eed (%) |
| a The reaction was performed with p-nitrobenzaldehyde (0.25 mmol), cyclohexanone (1.0 mmol), catalyst 1c (5 mg to 100 mg 0125 mmol g−1), in the presence of water (1.0 mL) at room temperature.b Isolated yield after chromatography on silica gel.c Determined by chiral 1HNMR analysis, major product is anti.d Determined by chiral HPLC analysis of the anti-product. | ||||||
| 1 | 1c | 100 | H2O (1 mL) | 99 | 85:15 |
93 |
| 2 | 1c | 80 | H2O (1 mL) | 99 | 80:20 |
92 |
| 3 | 1c | 50 | H2O (1 mL) | 99 | 78:22 |
93 |
| 4 | 1c | 40 | H2O (1 mL) | 98 | 84:16 |
92 |
| 5 | 1c | 30 | H2O (1 mL) | 98 | 90:10 |
94 |
| 6 | 1c | 20 | H2O (1 mL) | 98 | 94:6 |
96 |
| 7 | 1c | 10 | H2O (1 mL) | 98 | 98:2 |
99 |
| 8 | 1c | 5 | H2O (1 mL) | 90 | 98:2 |
95 |

Different results were observed with different amounts of water (Table 5), and the optimized amount of water was 1.0 mL. Finally, 10 mg of catalyst 1c employed in the presence of water (1.0 mL) was the optimized catalytic condition for the direct aldol reaction between cyclohexanone and p-nitrobenzaldehyde.
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | Cat. loading [%] | Water (mL) | Yieldb (%) | anti:sync |
eed (%) |
| a The reaction was performed with p-nitrobenzaldehyde (0.25 mmol), cyclohexanone (1.0 mmol), catalyst 1c (10 mg, 0.125 mmol g−1), in the presence of water at room temperature.b Isolated yield after chromatography on silica gel.c Determined by chiral 1HNMR analysis, major product is anti.d Determined by chiral HPLC analysis of the anti-product. | ||||||
| 1 | 1c | 10 | 0.2 | 94 | 91:9 |
94 |
| 2 | 1c | 10 | 0.4 | 92 | 89:11 |
94 |
| 3 | 1c | 10 | 0.5 | 93 | 91:9 |
94 |
| 4 | 1c | 10 | 0.8 | 91 | 90:10 |
94 |
| 5 | 1c | 10 | 1.0 | 98 | 98:2 |
99 |
| 6 | 1c | 10 | 1.5 | 85 | 91:9 |
92 |

To widen the range of substrates, we investigated ketone as an aldol donor with a series of aldehydes using catalyst 1c in the presence of water at room temperature under the optimized reaction conditions. As revealed in Table 6, the processes proceeded smoothly with 10 mg (0.5 mol%, 0.125 mmol g−1) of catalyst 1c resulting in highly enantio-enriched adducts in good yields regardless of the electronic nature of the aromatic aldehydes. For the neutral and electron-rich aromatic aldehydes, a longer reaction time was required compared to that required for the electron-deficient aromatic aldehydes. However, the aldol process proceeded efficiently in good yield (99%) with high enantioselectivity (99%). The good performance of heterogeneous catalyst 1c with regard to the ee value was most likely due to its novel support and good hybridization properties of the inorganic and organic components. The microenvironment of ZrPS-PVPA-Pr for asymmetric aldol reactions consisted of hydrophobic polystyrene potions and hydrophilic hybrid zirconium phosphonate portions with a self-assembled layered structure on the nanometre scale. Due to the interaction of a large number of polystyrene segments and layered zirconium phosphonate–phosphate existing in ZrPS-PVPA-Pr, a substantial number of caves, holes, pores and channels were formed due to folding, curling, parallelling, crossing, and even twining, which resulted in the excellent catalytic performance of 1c. However, this result is combined with the threshold effect assigned to the secondary channels, which have various sizes and shapes that are formed during the reaction in the presence of water.
|
|||||
|---|---|---|---|---|---|
| Entry | Product | Time (h) | Yieldb (%) | anti:sync |
eed (%) |
| a The reaction was performed with p-nitrobenzaldehyde (0.25 mmol), cyclohexanone (1.0 mmol), catalyst 1c (10 mg 0125 mmol g−1), in the presence of water (1.0 mL) at room temperature.b Isolated yield after chromatography on silica gel.c Determined by chiral 1HNMR analysis, major product is anti.d Determined by chiral HPLC analysis of the anti-product. | |||||
| 1 | 2(R = p-NO2-C6H4) | 24 | 98 | 98:2 |
99 |
| 2 | 3(R = o-NO2-C6H4) | 24 | 99 | 99:1 |
99 |
| 3 | 4(R = m-NO2-C6H4) | 24 | 97 | 97:3 |
99 |
| 4 | 5(R = p-CN-C6H4) | 24 | 98 | 98:2 |
98 |
| 5 | 6(R = p-CF3-C6H4) | 24 | 98 | 98:2 |
97 |
| 6 | 7(R = p-Br-C6H4) | 36 | 95 | 97:3 |
98 |
| 7 | 8(R = p-Cl-C6H4) | 36 | 96 | 98:2 |
99 |
| 8 | 9(R = o-Cl-C6H4) | 36 | 95 | 96:4 |
99 |
| 9 | 10(R = m-Cl-C6H4) | 36 | 92 | 94:6 |
98 |
| 10 | 11(R = p-F-C6H4) | 36 | 95 | 96:4 |
98 |
| 11 | 13(R = p-OMe-C6H4) | 36 | 84 | 93:7 |
96 |
| 12 | 14(R = m-OMe-C6H4) | 36 | 85 | 94:6 |
97 |
| 13 | 15(R = 2-naphthyl) | 36 | 86 | 94:6 |
94 |
| 14 | 16(R = 1-naphthyl) | 36 | 87 | 95:5 |
97 |
| 15 | 17(R = C6H5) | 36 | 85 | 92:8 |
93 |

To verify that catalyst 1c can be recovered and reused, we performed a recycling study of 1c using the aldol reaction between cyclohexanone and p-nitrobenzaldehyde (Table 7). Catalyst 1c was recovered by simple filtration, washed thoroughly with ethanol or water and dried at 60 °C to remove all of the impurities followed by direct use in a subsequent aldol reaction without the addition of any new catalyst. In each reuse cycle, the same amount of substrate was used, and without further purification, the recovered catalyst 1c retained its catalytic activity. In addition, only a slight decrease in the enantioselectivity was observed after 6 cycles (we did not continue beyond six cycles). In general, the layered zirconium phosphonate was relatively stable. However, the layered structure could be destroyed under some extreme conditions, such as basic solutions. However, the virgin layered structure, channels, holes and caves were roughly recovered in standing under aqueous phase conditions, which was helpful for self-assembling of the layered zirconium phosphonate. Therefore, these types of heterogeneous catalysts have the singular advantage of recyclability. The slight decrease in the activity for more cycles may be due to a physical loss during the recovery process and/or by a gradual degradation of the catalysts under the aqueous phase conditions and continuous stirring.
|
||||
|---|---|---|---|---|
| Entry | Time (h) | Yieldb (%) | anti:sync |
eed (%) |
| a Reaction conditions: cyclohexanone (100 mmol), aldehyde (25 mmol), catalyst 1c (1.0 g), water (100 mL) at room temperature.b The combined isolated yield of the diastereomers.c Determined by chiral 1HNMR analysis, major product is anti.d Determined by HPLC analysis of the anti product. | ||||
| 1 | 24 | 99 | 99:1 |
99 |
| 2 | 24 | 99 | 98:2 |
99 |
| 3 | 24 | 99 | 98:1 |
99 |
| 4 | 24 | 99 | 97:3 |
98 |
| 5 | 24 | 98 | 97:3 |
98 |
| 6 | 24 | 97 | 97:3 |
98 |

In addition, the large-scale test was performed with 1 mol of an aromatic aldehyde and 4 equivalents of cyclohexanone with, the same catalyst loading (10 g, 0.5 mol%) as in the experimental scale. The experiments were facilely carried out using the same procedure as that used for the experimental scale reactions. Based on the results summarized in Table 8, the enantioselectivities maintained the same level in the large-scale reactions.
|
|||||
|---|---|---|---|---|---|
| Entry | Product | Time (h) | Yieldb (%) | anti:sync |
eed (%) |
| a Reaction conditions: cyclohexanone (4000 mmol), aldehyde (1000 mmol), catalyst 1c (40 g), H2O (4000 mL) at room temperature.b The combined isolated yield of the diastereomers.c Determined by chiral HPLC analysis, major product is anti.d Determined by HPLC analysis of the anti product. | |||||
| 1 |  |
24 | 99 | 99:1 |
99 |
| 2 |  |
24 | 99 | 97:3 |
99 |
| 3 |  |
36 | 98 | 98:2 |
99 |
| 4 |  |
36 | 95 | 96:4 |
99 |
| 5 |  |
36 | 85 | 93:7 |
94 |

According to the above results (Table 3) where catalyst 1c exhibited better performance in the presence of water compared to that in organic solvents, the influence of the microenvironment effect of ZrPS-PVPA immobilized chiral molecule with an improved hydrophilic property was due to the large number of hydroxyl groups. The exciting phenomenon was also confirmed by the layered porous micro-structure shown in Fig. 2, which demonstrated that the organic groups in zirconium phosphonates were located on the surface of the layers, the interlamellar region and interlayer surface.
Both zirconium phosphates and zirconium phosphonates have excellent supermolecular intercalation performance. Some guests, such as amine and alkyl ethanol, can be easily intercalated into layered zirconium phosphate or zirconium phosphate–phosphonate in a polar solvent (e.g., water, ethanol or water/ethanol). According to the principle of intercalation chemistry, the interlayer distance of the zirconium phosphate or zirconium phosphate–phosphonate increased, and the layer floors were broadened in the polar solvent (i.e., water). Therefore, the heterogeneous catalyst with the layer floors of zirconium phosphates that can be distended in ZrPS-PVPA possess the same properties in the presence of water and can provide sufficient interspace for substrates to access to the catalytic active sites.
Conclusion
In this paper, a novel chiral phenoxy-modified organic–inorganic hybrid support-zirconium poly(styrene-phenylvinyl-phosphonate)phosphate has been used as a catalyst support for aldol reactions, and the results highlight the versatility of this class of organic–inorganic hybrid catalyst supports. In addition, the important effects of water as a solvent for the asymmetric aldol reactions using chiral heterogeneous catalysts 1c and 1d have been investigated and discussed. Enantiomeric excess up to 99% was obtained along with little loss of activity and enantioselectivity, and catalyst 1c can be efficiently used on a large-scale with the enantioselectivities of the anti-aldol reactions being maintained at the same level, which offers great potential for application in industry. It is easy to foresee important developments of similar systems based on insoluble ZrPS-PVPA. Our results also generate a new opportunity for understanding and designing organic–inorganic hybrid catalysts for chiral synthesis.Acknowledgements
The authors are grateful to National Natural Science Foundation of China (Grant no. 41203047) and Chongqing Unis Chemical Co, Ltd of China for financial support.References
- B. M. Trost, Science, 1991, 254, 1471–1477 CAS
.
- A. Corma, Chem. Rev., 1995, 95, 559–614 CrossRef CAS
- W. F. Hoelderich, Catal. Today, 2000, 62, 115–130 CrossRef CAS
- E. M. Carreira, in Comprehensive Asymmetric Catalysis, ed. E. N. Jacobsen, A. Pfaltz and H. Yamamoto, 1999, pp. 997–1065 Search PubMed
- J. G. Hernández and E. Juaristi, J. Org. Chem., 2011, 76, 1464–1467 CrossRef PubMed
- U. Eder, G. Sauer and R. Wiechert, Angew. Chem., Int. Ed., 1971, 10, 496–497 CrossRef CAS
- Z. G. Hajos and D. R. Parrish, J. Org. Chem., 1974, 39, 1615–1621 CrossRef CAS
- B. List, R. A. Lerner and C. F. Barbas III, J. Am. Chem. Soc., 2000, 122, 2395–2396 CrossRef CAS
- A. Cordova, W. Notz and C. F. Barbas III, Chem. Commun., 2002, 3024–3025 CAS
- S. Bahmanyar, K. N. Houk, H. J. Martin and B. List, J. Am. Chem. Soc., 2003, 125, 2475–2479 CrossRef CAS PubMed
- A. B. Northrup, I. K. Mangion, F. Hettche and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2004, 43, 2152–2154 CrossRef CAS PubMed
- Z. Tang, F. Jiang, X. Cui, L. Z. Gong, A. Q. Mi, Y. Z. Jiang and Y. D. Wu, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5755–5760 CrossRef CAS PubMed
- D. Ender and C. Grondal, Angew. Chem., Int. Ed., 2005, 44, 2 Search PubMed
- U. Kazmaier, Angew. Chem., Int. Ed., 2005, 44, 2186–2188 CrossRef CAS PubMed
- L. He, Z. Tang, L. F. Cun, A. Q. Mi, Y. Z. Jiang and L. Z. Gong, Tetrahedron, 2006, 62, 346–351 CrossRef CAS PubMed
- X. Y. Xu, Z. Tang, Y. Z. Wang, S. W. Luo, L. F. Cun and L. Z. Gong, J. Org. Chem., 2007, 72, 9905–9913 CrossRef CAS PubMed
- A. Russo, G. Botta and A. Lattanzi, Tetrahedron, 2007, 63, 11886–11892 CrossRef CAS PubMed
- L. S. Zu, H. X. Xie, H. Li, J. Wang and W. Wang, Org. Lett., 2008, 10, 1211–1214 CrossRef CAS PubMed
- Y. B. Liu, Q. Tong, L. Y. Ge, Y. Zhang, L. Hua and Y. N. Fan, RSC Adv., 2014, 4, 50412–50416 RSC
- S. P. Zhang, X. K. Fu, S. D. Fu and J. F. Pan, Catal. Commun., 2009, 10, 401–405 CrossRef CAS PubMed
- S. P. Zhang, X. K. Fu and S. D. Fu, Tetrahedron Lett., 2009, 50, 1173–1176 CrossRef CAS PubMed
- R. T. Baker and W. Tumas, Science, 1999, 284, 1477–1479 CrossRef CAS
- Q. H. Xia, H. Q. Ge, C. P. Ye, Z. M. Liu and K. X. Su, Chem. Rev., 2005, 105, 1603–1662 CrossRef CAS PubMed
- U. Costantino, F. Fringuelli, M. Nocchetti and O. Piermatti, Appl. Catal., A, 2007, 326, 100–105 CrossRef CAS PubMed
- S. J. Bae, S. W. Kim, T. Hyeon and B. M. Kim, Chem. Commun., 2000, 31–32 CAS
- H. M. L. Davies, C. Venkataramani, T. Hansen and D. W. Hopper, J. Am. Chem. Soc., 2003, 125, 6462–6463 CrossRef CAS PubMed
- B. M. Choudary, B. K. Avita, C. N. Sreenivasa, B. Sreedhar and K. M. Lakshmi, Catal. Lett., 2002, 78, 373–385 CrossRef CAS
- M. Benaglia, M. Cinquini, F. Cozzi, A. Puglisi and G. Celentano, Adv. Synth. Catal., 2002, 344, 533–542 CrossRef CAS
- Y. Y. Wu, Y. Z. Zhang, M. L. Yu, G. Zhao and S. W. Wang, Org. Lett., 2006, 8, 4417–4420 CrossRef CAS PubMed
- T. M. Suzuki, S. Kobayashi, D. A. Pacheco Tanaka, M. A. Llosa Tanco, T. Nagase and Y. Onodera, React. Funct. Polym., 2004, 58, 131–135 CrossRef CAS PubMed
- A. A. Marti and J. L. Colon, Inorg. Chem., 2003, 42, 2830–2832 CrossRef CAS PubMed
- U. Costantino, M. Nocchetti and R. Vivani, J. Am. Chem. Soc., 2002, 124, 8428–8432 CrossRef CAS PubMed
- W. S. Ren and X. K. Fu, Catal. Commun., 2009, 10, 788–793 CrossRef CAS PubMed
- I. C. Marcu, I. Sandulescu and J. M. M. Millet, Appl. Catal., 2002, 227, 309–320 CrossRef CAS
- S. Calogero, D. Lanari, M. Orru, O. Piermatti, F. Pizzo and L. Vaccaro, J. Catal., 2011, 282, 112–119 CrossRef CAS PubMed
- M. Angeloni, O. Piermatti, F. Pizzo and L. Vaccaro, Eur. J. Org. Chem., 2014, 1716–1726 CrossRef CAS
- X. J. Wu, X. B. Ma, Y. L. Ji, Q. Wang, X. Jia and X. K. Fu, J. Mol. Catal. A: Chem., 2007, 265, 316–322 CrossRef CAS PubMed
- P. A. Grieco, Organic Synthesis in Water, Blackie Academic & Profesional: London, 1998 Search PubMed
- J. M. Huang, X. T. Zhang and D. W. Armstrong, Angew. Chem., Int. Ed., 2007, 46, 9073–9077 CrossRef CAS PubMed
- J. G. Hernández and E. Juaristi, Chem. Commun., 2012, 48, 5396–5409 RSC
- C. L. Wu, X. K. Fu, X. B. Ma and S. Li, Tetrahedron: Asymmetry, 2010, 21, 2465–2470 CrossRef CAS PubMed
- C. L. Wu, X. K. Fu and S. Li, Eur. J. Org. Chem., 2011, 1291–1299 CrossRef CAS
- C. L. Wu, X. K. Fu and S. Li, Tetrahedron, 2011, 67, 4283–4290 CrossRef CAS PubMed
- C. L. Wu, X. K. Fu and S. Li, Tetrahedron: Asymmetry, 2011, 22, 1063–1073 CrossRef CAS PubMed
- C. L. Wu, X. Q. Long, S. Li and X. K. Fu, Tetrahedron: Asymmetry, 2012, 23, 315–328 CrossRef CAS PubMed
- S. Li, C. L. Wu, X. K. Fu and Q. M, Ind. Eng. Chem. Res., 2011, 50, 13711–13716 CrossRef CAS
- B. W. Gong, X. K. Fu, J. X. Chen, Y. D. Li, X. C. Zou, X. B. Tu, P. P. Ding and L. P. Ma, J. Catal., 2009, 262, 9–17 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: General experimental methods and spectra of the corresponding compounds. See DOI: 10.1039/c4ra11208c |
| This journal is © The Royal Society of Chemistry 2015 |