
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNitric oxide release by N-(2-chloroethyl)-N-nitrosoureas: a rarely discussed mechanistic path towards their anticancer activity†
Amrita
Sarkar‡
,
Subhendu
Karmakar‡
,
Sudipta
Bhattacharyya
,
Kallol
Purkait
and
Arindam
Mukherjee
*
Department of Chemical Sciences, Indian Institute of Science Education & Research Kolkata, Mohanpur, Nadia, Pin – 741246, West Bengal, India. E-mail: a.mukherjee@iiserkol.ac.in; Fax: +91-033-25873020; Tel: +91-033-25873031
First published on 28th November 2014
Abstract
N-(2-Chloroethyl)-N-nitrosoureas (viz. carmustine (BCNU), lomustine) are used in critical cases of brain tumors or leukemia since these compounds are very toxic in nature. The mechanistic pathways of these compounds project the generation of isocyanates along with [–N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N–CH2–CH2–Cl]+OH− which, due to its reactivity, will breakdown further to produce DNA adducts and various other metabolites. Almost all of the literature predicting the mechanism does not discuss nitric oxide (NO) release as one of the viable mechanistic pathways for these N-(2-chloroethyl)-N-nitrosoureas. Our studies with two aromatic N-(2-chloroethyl)-N-nitrosoureas show that NO release may be a facile pathway for their activity. We have probed the NO release through fluorescence and the traditional Griess reagent assay. The aqueous stability studies show that the rates of decomposition of the two aromatic N-(2-chloroethyl)-N-nitrosoureas are of the order of 10−2 min−1. Initial studies of the cytotoxicity against two different cancer cell lines show that they are quite efficient even under hypoxic conditions, compared to the clinical compound BCNU, based on the results on human breast (MCF-7) and lung (A549) adeno carcinoma cell lines. The IC50 values range from about 38–95 μM. Encouragingly the 1-(4-chloro-1,2-phenylene)bis(3-(2-chloroethyl)-3-nitrosourea) (2a) is selectively more toxic to the A549 cancer cells (IC50 = 41 ± 4 μM) than the normal mouse embryonic fibroblast (NIH3T3, IC50 63 ± 4 μM) or non-tumorigenic human embryonic kidney cell line (HEK293T, IC50 = 83 ± 2 μM) and arrests the cell cycle in the G2/M-phase in the A549 cell line. Even under hypoxic conditions compound 2a is active at 49 ± 5 μM.
N–CH2–CH2–Cl]+OH− which, due to its reactivity, will breakdown further to produce DNA adducts and various other metabolites. Almost all of the literature predicting the mechanism does not discuss nitric oxide (NO) release as one of the viable mechanistic pathways for these N-(2-chloroethyl)-N-nitrosoureas. Our studies with two aromatic N-(2-chloroethyl)-N-nitrosoureas show that NO release may be a facile pathway for their activity. We have probed the NO release through fluorescence and the traditional Griess reagent assay. The aqueous stability studies show that the rates of decomposition of the two aromatic N-(2-chloroethyl)-N-nitrosoureas are of the order of 10−2 min−1. Initial studies of the cytotoxicity against two different cancer cell lines show that they are quite efficient even under hypoxic conditions, compared to the clinical compound BCNU, based on the results on human breast (MCF-7) and lung (A549) adeno carcinoma cell lines. The IC50 values range from about 38–95 μM. Encouragingly the 1-(4-chloro-1,2-phenylene)bis(3-(2-chloroethyl)-3-nitrosourea) (2a) is selectively more toxic to the A549 cancer cells (IC50 = 41 ± 4 μM) than the normal mouse embryonic fibroblast (NIH3T3, IC50 63 ± 4 μM) or non-tumorigenic human embryonic kidney cell line (HEK293T, IC50 = 83 ± 2 μM) and arrests the cell cycle in the G2/M-phase in the A549 cell line. Even under hypoxic conditions compound 2a is active at 49 ± 5 μM.
Introduction
N-(2-chloroethyl)-N-nitrosoureas represent a class of compounds with a wide range of activities mainly used against leukaemia and brain tumors.1–6 Carmustine and lomustine (Scheme 1) are two such FDA approved drugs belonging to this class and are mainly used in leukaemia and multiple myeloma.7,8 Fotemustine and nimustine (Scheme 1) are newer drugs in the same category under clinical trials.9–13 The varied use of N-(2-chloroethyl)-N-nitrosoureas shows that although they are of the same family, their pathway of action may be different. All the clinically used nitrosourea drugs show haematological toxicity.14–16 Following the evolution of N-nitrosourea drugs, it was found that from simple aliphatic, alicylic, aromatic or heterocyclic compounds the focus gradually shifted to more complex compounds conjugated with biorelevant carrier moieties viz. amino acids, carbohydrates, nucleosides, steroids etc. to achieve better toxicity along with specificity towards cancer cells.3,7,17–27 | ||
| Scheme 1 Different nitrosoureas as chemotherapeutic agents. | ||
Although nitrosourea contain a nitric oxide (NO) group, the dissociation pathways shown mostly does not include the proposition of NO release so far except for the work of Lown et al. which although does not show any direct evidence of NO release but through detection of other intermediates has discussed NO release as one of the pathways.28–30 The mechanism of action of nitrosoureas are known to be complex and various species are postulated on direct or indirect evidences.4,28,29,31–34 Most of the mechanisms proposed stresses mainly upon the formation of isocyanates, [–NN–CH2–CH2–Cl]+OH− and +CH2–CH2–Cl in addition to various other minor species but no NO release is generally reported except in two cases28,30 where the authors argued in favour of NO release as one of the pathways. Except for those two isolated reports the other studies support the formation of HO–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–CH2–CH2–Cl, and +CH2–CH2–Cl intermediates.2,31,33,35–38
N–CH2–CH2–Cl, and +CH2–CH2–Cl intermediates.2,31,33,35–38
Our studies presented here show that NO release seems to be a facile and viable mechanistic pathway for aromatic N-(2-chloroethyl)-N-nitrosoureas. Ever since the mechanism of endogenous nitric oxide (NO) was established in 1982 it has been extensively probed for various biomedical applications.39–51 It is known that NO at higher doses (exceeding 1 μM) trigger cell cycle arrest52 thus NO donors may be potent anticancer agents.42,53–55
We have synthesized two aromatic N-(2-chloroethyl)-N-nitrosourea (Scheme 2) to study their stability profile, NO release ability and attempt to correlate the aqueous decomposition and NO release ability to their cytotoxic activity towards cancer cells. Our target compounds were 1a and 2a out of which the synthetic procedure for 1a was reported earlier but the compound was not studied for aqueous stability, NO release or in vitro cytotoxicity.17,561 and 2 were good precursors to perform comparative studies with their nitroso derivatives, 1a and 2a whenever needed.
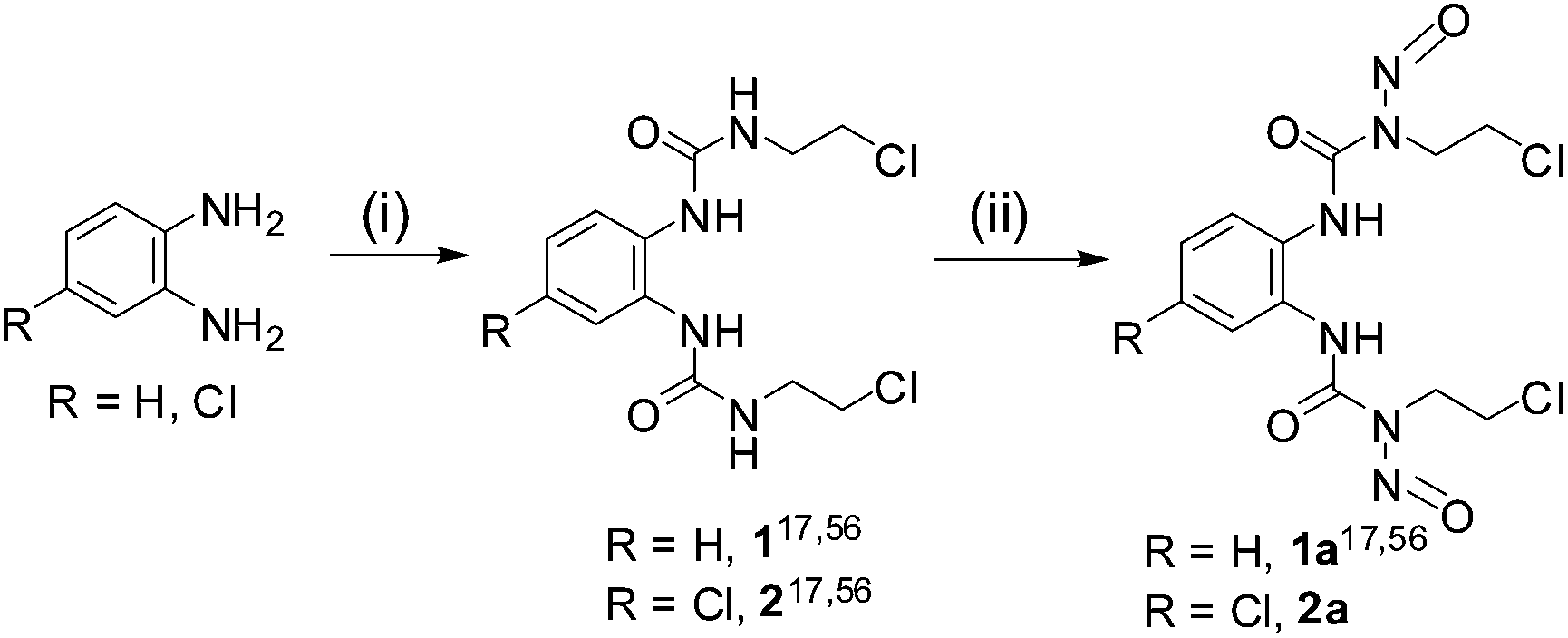 | ||
| Scheme 2 Representative scheme for synthesis of nitrosoureas (1a, 2a) (where 1, 2 and 1a were synthesized according to literature procedure17,56). (i) 2-Chloroethyl isocyanate (2 equiv.), DCM/THF, 0 °C, 0 °C to RT, 6 h (ii) NaNO2–HCO2H, 0–5 °C, 4 h. | ||
Results and discussion
Syntheses and characterization
The synthesis of ureas (1, 2), nitrosoureas (1a, 2a) proceeded as depicted in Scheme 2. 1,1′-(1,2-phenylene)bis(3-(2-chloroethyl)urea) (1), 1-(4-chloro-1,2-phenylene)bis(3-(2-chloroethyl)urea) (2) and 1,1′-(1,2-phenylene)bis(3-(2-chloroethyl)-3-nitrosourea) (1a) were prepared using earlier reported literature procedure.17,56 The aromatic 2-chloroethylurea derivatives were synthesized in one step using a isocyanate and the nitroso derivatives were synthesized in a single step using NaNO2–HCOOH.17,56 Hence, using two well defined synthetic steps we can synthesize 1a and 2a from commercial precursors. The characterization using NMR showed that the synthesized compounds were pure. The aromatic protons of 1 and 1a are multiplets instead of doublets (details in Experimental section). This may be assigned to the difference in spatial orientation of the N-(2-chloroethyl)-N-nitrosourea moieties reducing the symmetry of the molecule as found in the crystal structure of 1a. The analytical purity of the bulk products were further confirmed by CHN analysis. The ESI-MS of the compounds are also in good agreement with proposed molecular formulation.Structural description
The compound 1a (Fig. 1) was characterized by single crystal X-ray crystallography. It crystallizes in triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . Single crystals suitable for X-ray crystallography were grown by keeping a saturated DMSO solution at 20 °C for a day. The important crystallographic parameters and selected bond length and angles have been summarised in Tables S1 and S2 (ESI†) respectively. The molecular structure of 1a revealed that C–N bonds in urea group are asymmetric. The C(7)–N(4) adjacent to phenyl ring has a bond distance of 1.340(3) Å whereas the same for C(7)–N(5), adjacent to the nitroso group, is 1.424(3) Å showing the influence of the phenyl ring on the double bond character. The N–O bond lengths of 1.223(2) Å and 1.224(3) Å are in good agreement with earlier reported N-nitrosourea monomer.57,58 The molecule has a intramolecular hydrogen bonding (2.729 Å) between O(3) and N(1) and intermolecular hydrogen bonding between O(1) of one molecule to N(4) of a neighbouring molecule.
. Single crystals suitable for X-ray crystallography were grown by keeping a saturated DMSO solution at 20 °C for a day. The important crystallographic parameters and selected bond length and angles have been summarised in Tables S1 and S2 (ESI†) respectively. The molecular structure of 1a revealed that C–N bonds in urea group are asymmetric. The C(7)–N(4) adjacent to phenyl ring has a bond distance of 1.340(3) Å whereas the same for C(7)–N(5), adjacent to the nitroso group, is 1.424(3) Å showing the influence of the phenyl ring on the double bond character. The N–O bond lengths of 1.223(2) Å and 1.224(3) Å are in good agreement with earlier reported N-nitrosourea monomer.57,58 The molecule has a intramolecular hydrogen bonding (2.729 Å) between O(3) and N(1) and intermolecular hydrogen bonding between O(1) of one molecule to N(4) of a neighbouring molecule.
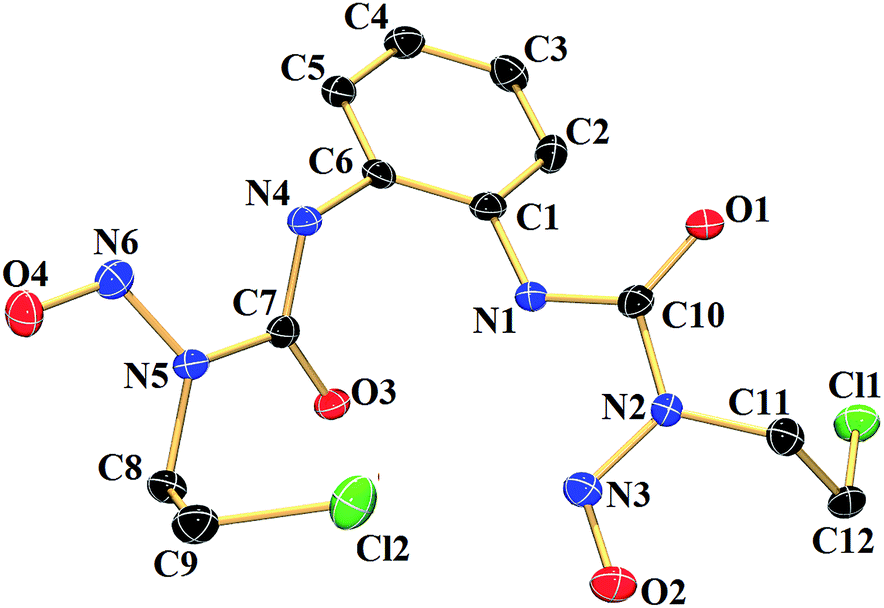 | ||
| Fig. 1 Molecular structure of 1a with 50% probability of thermal ellipsoids. Hydrogen atoms have been omitted for clarity. | ||
Stability studies
The half lives of the N-nitrosoureas were monitored via UV-vis absorption spectroscopy in buffer at two different pH, 7.4 and 6.0. Tumour cells have hypoxic regions which may have relatively lower interstitial pH due to the accumulation of metabolic products from glycosis.59,60 Since the decomposition may be pH dependent, more decomposition at lower pH may lead to the compound being degraded faster which may prohibit its entry inside a cell. Hence the compounds were studied for their decomposition at pH 6.0. The decrease in the absorption maxima around 196–242 nm of the compounds (1a, 2a and nimustine hydrochloride) were monitored with time. The rate of aqueous decomposition (kD) and corresponding half life (t1/2) were calculated by plotting the ln(Abs) values vs. time plot. It was observed from the aqueous decomposition rate for both 1a and 2a that the rate of 1a is comparatively faster than 2a. Both 1a and 2a were found to be decomposing faster than nimustine hydrochloride (Fig. 2). In lower pH (pH = 6.0) in every case there is a decrease in aqueous decomposition rate although the difference is less (Table 1). The absorbance vs. wavelength plots were shown in Fig. S1–S3 (ESI†) and the inset figures show corresponding ln(Abs) vs. time plot. The nitrosoureas 1a and 2a are reactive due to the presence of the NO group. The rate of decomposition at pH 7.4 shows the order is 1a > 2a (Table 1). 1a decompose faster than 2a and the only difference in the formulation of 2a is the presence of the chloroaryl moiety instead of aryl in 1a (Scheme 2). Hence the +R effect of the chloro group is responsible for slowing the aqueous decomposition rate.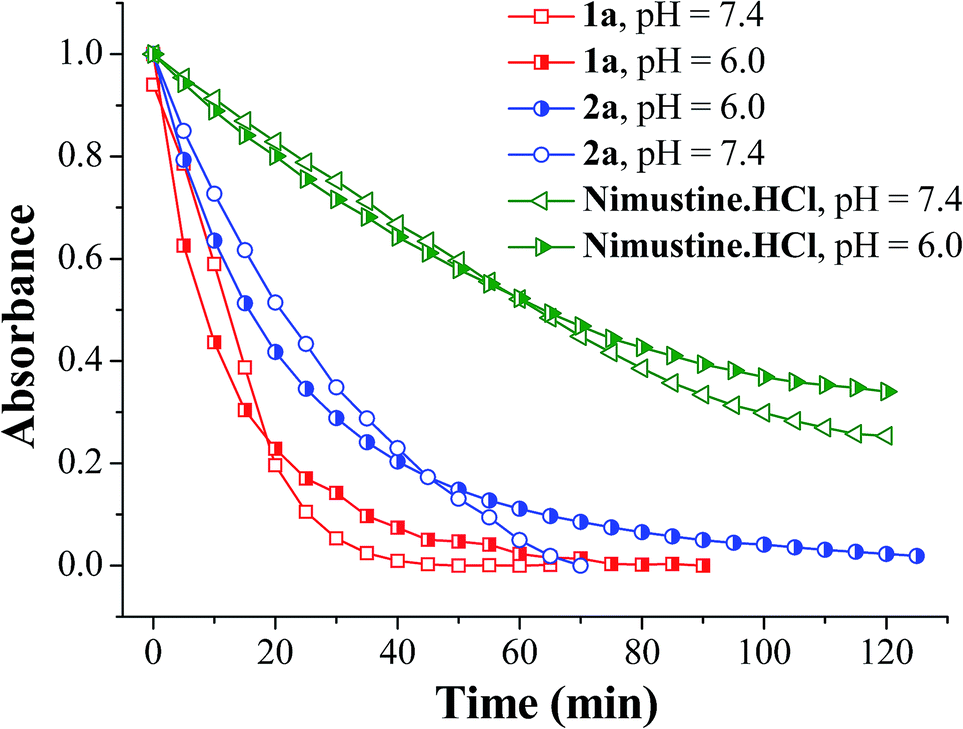 | ||
| Fig. 2 Decrease in absorbance (normalised) vs. time plot for aqueous stability of compounds 1a, 2a and nimustine HCl in phosphate buffer (10 mM) of pH 7.4 and 6.0. The decrease in absorbance at λ = 236 nm for 1a, 242 nm for 2a, and 232 nm for nimustine HCl with time has been monitored and plotted. | ||
The UV spectrum also shows multiple isobestic point (Fig. S1–S3, ESI†) indicating that there are two different species in solution, which are UV active and one is forming steadily from the other. From an apparent look it would appear that nimustine which is a N-(2-chloroethylnitrosourea) class compound and is in clinical trial has decomposition rate much slower than that of 1a or 2a (Table 1). However, it should be noted that per molecule there are two N-(2-chloroethyl)-N-nitrosourea moieties in 1a and 2a in contrast to one in nimustine. Accounting for this fact it shows that the aqueous decomposition rate per N-(2-chloroethyl)-N-nitrosourea moiety is ca. 2.81 × 10−2 for 1a and 8.80 × 10−3 min−1 for 2a. We found that at pH 6.0 the rate of decomposition slows down and the rate is 1.72(9) × 10−2, 1.20(4) × 10−2 and 9.73(3) × 10−4 min−1 for 1a, 2a and nimustine respectively (Table 1). Hence, it shows that when the pH is acidic (pH 6.0), the reduction in decomposition rate is more pronounced for nimustine as compared to 1a and 2a. In fact the decomposition rate for compound 2a is similar in both hypoxia and normoxia. Hence, the activity of 2a in hypoxia and normoxia may not show much difference. 1a and 2a have two dissociable N-(2-chloroethyl)-N-nitrosourea moieties per molecule which is also more atom economic compared to nimustine since there would be dissociation of one arene by product per two alkylating moiety and NO released. One would expect that these aqueous decomposition data would show correlation with the anticancer activity.
Potency to inhibit cancer cells in vitro
All the four compounds were probed in vitro against human breast adenocarcinoma cell line (MCF-7), human lung adenocarcinoma epithelial cell line (A549), non tumerogenic human embryonic kidney cell (HEK293T) and mouse embryonic fibroblast cell line (NIH3T3) by MTT assay and cisplatin was used as a standard to check the quality of data in each 96 well plate. Two well known compounds belonging to N-nitrosourea family BCNU (FDA approved drug) and nimustine (under clinical trials) were also tested to evaluate their corresponding IC50 values under same conditions in MCF-7 and A549, HEK293T and NIH3T3 cell lines. The data are summarized in Table 2 and corresponding IC50 plots of 1a and 2a in different cancer cell lines using GraphPad Prism 5® are given in Fig. S4–S9, (ESI†). The hypoxic activation was also probed by cytotoxicity assay of 1a and 2a in hypoxic conditions using oxygen percentage of 1.5%. Both the nitrosourea compounds are more active in A549 as compared to MCF-7 (Table 2). Compound 1a and 2a have similar cytotoxicity (IC50 = 38 ± 1 μM for 1a and 41 ± 5 for 2a) in A549 under normoxic condition. 1a bearing the N-(2-chloroethyl)-N-nitrosourea without the chloro in the aryl moiety, has the fastest rate of decomposition and showed better activity (Table 2). In both A549 and MCF-7 cell line 1a and 2a show similar activity in normoxia and hypoxia (Table 2). The MTT assay of BCNU (a clinical drug) and nimustine under same conditions as our compounds (details in Experimental section) does not show any significant killing (survival > 75%) up to 150 μM in normoxia or hypoxia. We also tested 1a and 2a in a non tumerogenic (HEK293T) and a primary cell line (NIH3T3). Primary cell lines are generally difficult to culture in vitro conditions yet the IC50 values suggest that 2a is much less toxic to the HEK293T and NIH3T3 as compared to its activity in A549 cell line. 1a was less toxic for HEK293T but showed similar toxicity in NIH3T3 when compared with A549.| Compounds | IC50a (μM) ± SD | |||||
|---|---|---|---|---|---|---|
| MCF-7 | A549 | HEK293T | NIH3T3 | |||
| Normoxia | Hypoxia | Normoxia | Hypoxia | Normoxia | Normoxia | |
| a IC50 = concentration required to effect 50% inhibition of cell growth, values are means ± standard deviation of three independent experiments carried out by triplicates, p < 0.05. IC50 values were calculated by variable slope model using GraphPad Prism 5®. Cells (6 × 103 per well) were treated with increasing concentrations of compounds for 48 h. b At 150 μM survival rate was 75–80%. c n.d = not determined. | ||||||
| 1 | >500 | n.dc | >500 | n.d | n.d | n.d |
| 2 | 244 ± 1 | n.d | >350 | n.d | n.d | n.d |
| 1a | 95 ± 2 | 89 ± 2 | 38 ± 3 | 40 ± 2 | 86 ± 2 | 39 ± 3 |
| 2a | 90 ± 3 | 86 ± 1 | 41 ± 4 | 49 ± 5 | 83 ± 2 | 63 ± 4 |
| BCNU | >150b | >150b | >150b | >150b | >150b | >150b |
| Nimustine HCl | >150b | >150b | >150b | >150b | >150b | >150b |
| Cisplatin | 14 ± 1 | 19 ± 1 | 22 ± 2 | 24 ± 2 | 9 ± 2 | 6 ± 2 |
We also performed some initial studies on the cell cycle inhibition by flow cytometry (FACS) with 2a. After 24 hours of incubation with 2a the respective DNA content were analysed by FACS (Table S3†). The results showed that compound 2a lead to arrest of A549 cells in G2/M phase (Fig. 3 and S10, ESI†) signifying that those cells where there has been significant DNA damage could not enter the next phase M, of the cell cycle, and if somehow they have escaped the G2 phase arrest, then the cells were trapped in M phase. The optical microscopy image data of A549 cells after 24 h treatment of 2a also support the cell cycle arrest data as visible change in nuclear morphology (nuclear elongation) was observed (Fig. S11†).
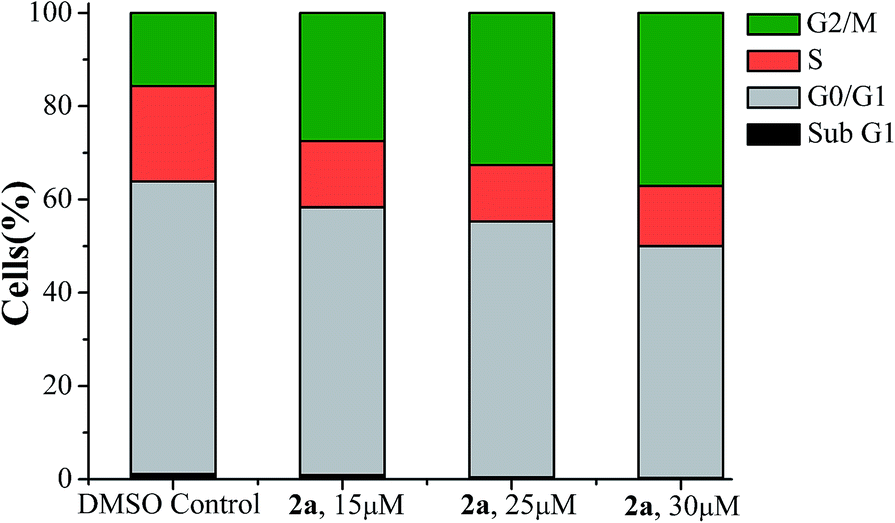 | ||
| Fig. 3 Cell cycle analysis of A549 cells after 24 h exposer to 2a in three different concentrations 15 μM, 25 μM and 30 μM respectively. | ||
Nitric oxide release studies
In order to monitor if release of nitric oxide (NO) is possible from the N-(2-chloroethyl)-N-nitrosoureas 1a and 2a Griess reagent test was performed (it should be noted that Griess reagent detects NO2− which is formed from NO by reaction with oxygen).52,61 The NO release reactions were performed by dissolving 1a, 2a and nimustine in a sealed vial in DMSO–water (7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 v/v) and after 5 min injecting the head space gas in to the Griess reagent vial. The nitrite detected was the oxidised product of nitric oxide in the reaction with Griess reagent (Scheme S1†) producing increase of absorbance at ∼540 nm. The test was found to be positive by development of the purple coloration. This was done over a period of time and the purple colouration was found to increase confirming the release of NO (Fig. 4). Since same amount of overhead gas from each sample was used the data obtained may show the trend in NO release although it would not be able to quantitate. The amount of NO released is of the order nimustine > 1a > 2a. Later we performed the same experiment using the traditional solution phase test52,61 (Fig. S12 & S13, ESI†) and found the same trend as obtained using the overhead gas. The rate constants (kNO) were calculated from the slope of ln(Abs) vs. time plots (Fig. S14, ESI†) and the rate constant (kNO) falls in the order, nimustine > 1a > 2a. Release of labile NO was confirmed and estimated by the increase of absorbance produced by the resultant dye at ∼540 nm. The estimated NO release from 1a, 2a and nimustine hydrochloride were compared with the data obtained from a well known nitrite donor compound, sodium nitrite, under the same experimental condition. Both 1a and 2a took ca. 4 h for achieving saturation but for nimustine it took much less time ca. 132 min (Fig. S12, ESI†).
:3 v/v) and after 5 min injecting the head space gas in to the Griess reagent vial. The nitrite detected was the oxidised product of nitric oxide in the reaction with Griess reagent (Scheme S1†) producing increase of absorbance at ∼540 nm. The test was found to be positive by development of the purple coloration. This was done over a period of time and the purple colouration was found to increase confirming the release of NO (Fig. 4). Since same amount of overhead gas from each sample was used the data obtained may show the trend in NO release although it would not be able to quantitate. The amount of NO released is of the order nimustine > 1a > 2a. Later we performed the same experiment using the traditional solution phase test52,61 (Fig. S12 & S13, ESI†) and found the same trend as obtained using the overhead gas. The rate constants (kNO) were calculated from the slope of ln(Abs) vs. time plots (Fig. S14, ESI†) and the rate constant (kNO) falls in the order, nimustine > 1a > 2a. Release of labile NO was confirmed and estimated by the increase of absorbance produced by the resultant dye at ∼540 nm. The estimated NO release from 1a, 2a and nimustine hydrochloride were compared with the data obtained from a well known nitrite donor compound, sodium nitrite, under the same experimental condition. Both 1a and 2a took ca. 4 h for achieving saturation but for nimustine it took much less time ca. 132 min (Fig. S12, ESI†).
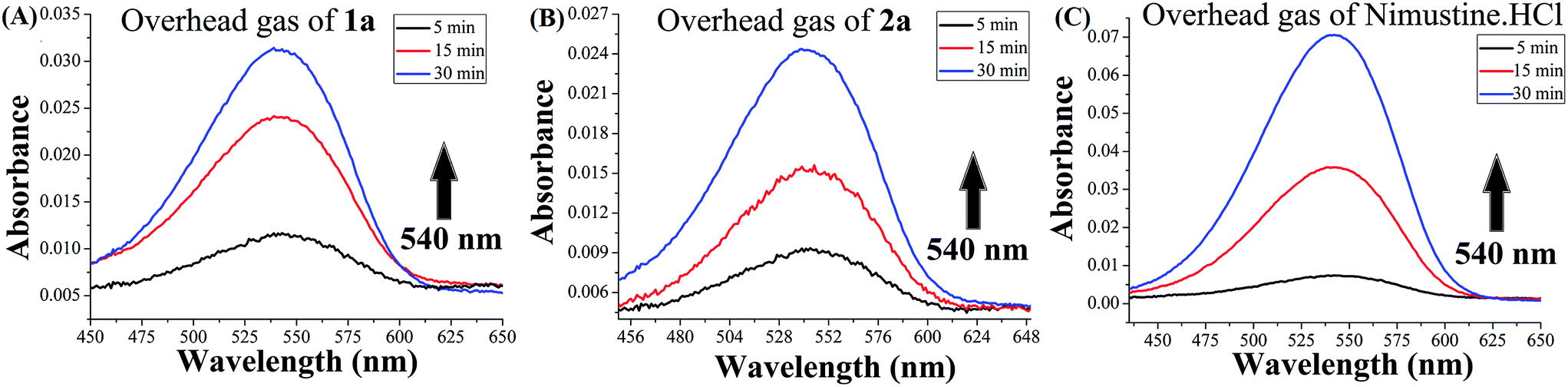 | ||
| Fig. 4 Change in absorbance vs. wavelength (λmax = 540 nm) when headspace gas from (A) 1a and (B) 2a and (C) nimustine HCl giving positive Griess reagent test. | ||
However, it should be noted that the solution phase (Table 3) NO release rates were under highly acidic conditions in solution (see Experimental section for details) which is different from normal physiology or cancer cells. Hence, the quantitation of NO release is only indicative and performed to compare the maximum NO release ability of 1a and 2a with nimustine. Compound 1 and 2 gave no colouration in Griess reagent assay under the experimental conditions indicating that there may not be an alternate reactive species that can mislead the Griess test. The NO release by nimustine or other aromatic N-(2-chloroethyl)-N-nitrosoureas viz.1a, 2a, is not known as per the literature. Out of the numerous literatures on detailed mechanistic pathways2,31,33,36–38 of the N-(2-chloroethyl)-N-nitrosoureas only the work of Lown et al.28–30 indicates NO release by BCNU, MeCCNU and CCNU as one of the possible pathways based on the intermediates formed and detected, due to NO release. However, no NO release experiment was reported. Our results also strongly agree with the NO release pathway and we have direct evidence of NO release by using the overhead gas for 1a, 2a and nimustine in Griess reagent assay.
| Compounds | [NO]t (μM) | k NO (min−1) | t 1/2 (min) | t d (min) | NO released (mol%) |
|---|---|---|---|---|---|
| a Compound concentration (1a, 2a and nimustine HCl) = 62.5 μM. [NO]t = total NO release, kNO = rate of NO release, t1/2 = half life of NO release, td = duration of NO release. All the above data are mean of two independent experiments. | |||||
| 1a | 25.5 | 5.88 × 10−3 | 118 | 230 | 20.4 |
| 2a | 23.5 | 4.19 × 10−3 | 165 | 234 | 18.8 |
| Nimustine HCl | 42.9 | 1.48 × 10−2 | 47 | 132 | 68.6 |
The detection of released nitric oxide from N-(2-chloroethyl)-N-nitrosourea (1a) as well as nimustine was also confirmed by fluorescence studies. Among various fluorescent probes available aromatic ortho-diamino compounds are most commonly used.62–64 1,2-Diamino anthraquinone (DAA) may be used as a sensitive and specific NO probe.65 1,2-Diamino compounds react with nitric oxide (NO) in presence of O2 to produce a fluorescent triazole derivative (DAA-Tz) (Scheme S2, ESI†) with a new emission maxima.65 The emission spectra of DAA in DMSO (50 μM) exhibit peak maxima at 638 nm with a weak shoulder at 570 nm (Fig. 5A). Upon treatment 50 μM of 1a the solution exhibits a new band at 562 nm overlapping with a band at 640 nm showing that both DAA and DAA-Tz exist in the solution (Fig. 5B). When we added the headspace gas (20 mL) generated from compound 1a the formation of triazole derivative (DAA-Tz) was confirmed by the peak at 574 nm (Fig. 5C). For nimustine hydrochloride the peak appeared at 571 nm was the confirmation of formation of triazole DAA-Tz (Fig. 5D). We have also compared the fluorescence of DAA-Tz formed by a known nitrosating agent NOBF4 which further supports that nitric oxide is releasing from N-(2-chloroethyl)-N-nitrosourea class of compounds studied here.
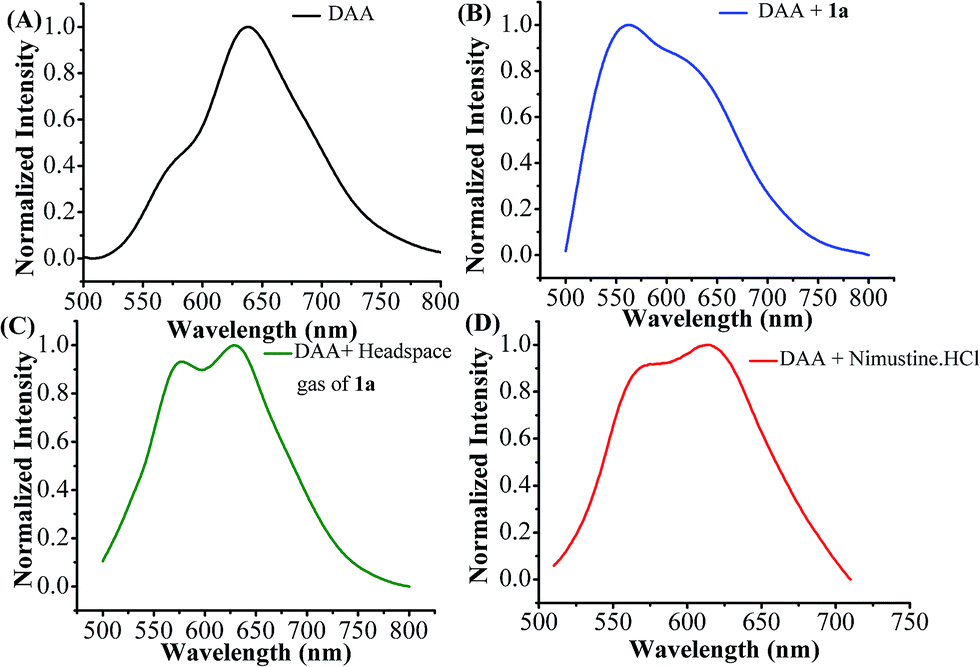 | ||
| Fig. 5 (A) DAA (50 μM in DMSO): normalized emission spectrum, λex = 490 nm. (B) DAA (50 μM in DMSO) + 1a (50 μM in DMSO): normalized emission spectrum, λex = 490 nm. (C) DAA (50 μM in DMSO) + headspace gas of 1a (20 mL): normalized emission spectrum, λex = 490 nm. (D) DAA (50 μM in DMSO) + nimustine HCl (50 μM in DMSO): normalized emission spectrum, λex = 490 nm. | ||
Our investigation to probe the NO release by probing the overhead gas as well as the solution of 1a and 2a using the traditional Griess reagent assay and by fluorescence spectroscopy forming DAA-triazole showed that NO indeed gets released from these compounds (Fig. 4, 5 and Table 3). Based on the above results we have proposed an alternate mechanism to show the nitric oxide release in Scheme 3. Apart from the evidence of NO release the mechanistic proposal was based on the mass spectrometry of solutions of 1a performed in 2:1 v/v MeCN–H2O mixture (2a being similar to 1a only the latter was studied through ESI-MS). We found that ESI-MS data shows the presence of imidazolone and isocyanates as the major products (Fig. S17A & S17B, ESI†) of aqueous decomposition. We also found existence of oxadiazolium cation in the ESI-MS of 1a (Scheme 3, Fig. S17A & S17B, ESI†). Hence we proposed the mechanistic pathway as depicted in Scheme 3, where path A shows the formation of oxadiazolium cation.
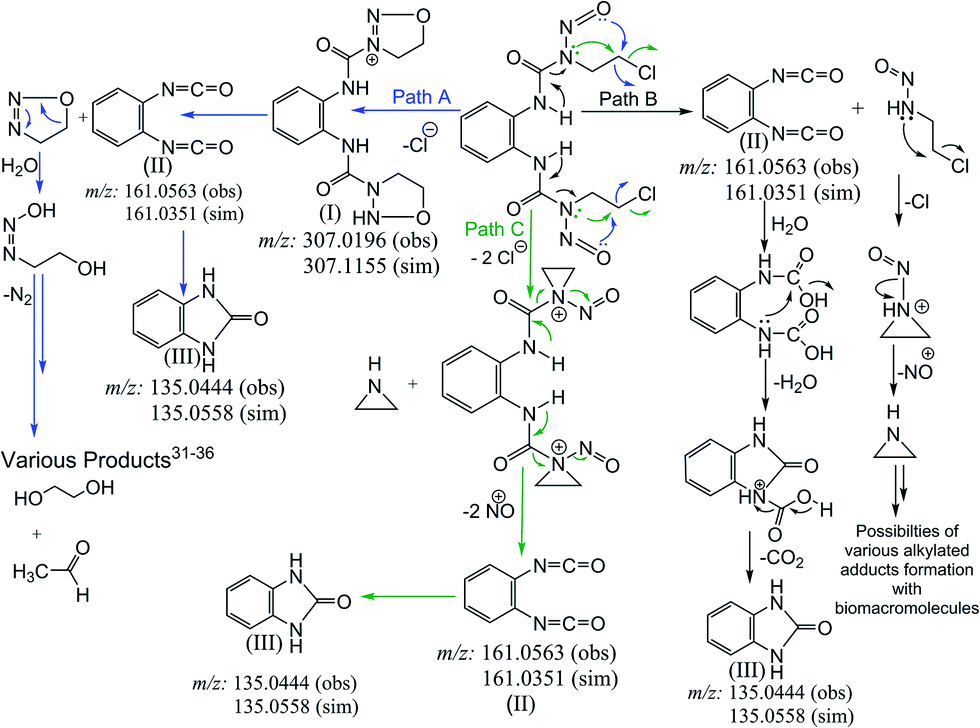 | ||
| Scheme 3 Schematic representation of proposed mechanistic pathways of N-nitrosourea (1a). | ||
The proposed pathways show that the major end products may be similar whether NO is released or not. We propose that NO release can happen at different stages depending on the compound and pathway followed (Scheme 3, Fig. S17A & S17B, ESI†). The reason behind this proposition is that the aqueous decomposition and NO release are not the same as found through our results. 1a and 2a show a higher decomposition rates and higher cytotoxicity but their NO release rate is slower. Nimustine on the other hand shows a slower decomposition rate but the NO release rate is higher even at pH 7.4 as per the overhead gas studies. However, Nimustine shows a poor in vitro cytotoxicity profile. Hence, it appears that the rate of decomposition does not provide indication of released NO. It appears that if NO is released at a faster rate than the cytotoxicity of the compound becomes poor which may be due to poor permeability of the resultant compound. Our ESI-MS studies of decomposition of BCNU and nimustine supports that the dissociation pathways are different (Schemes S3, S4 and Fig. S18, S19†). In case of 1a based on the NO detection and the ESI-MS data only path A and B are followed (Scheme 3). Based on the differences in the data it can be said that the NO release do not happen through the same pathway in nimustine and 1a or 2a. In case of nimustine the percentage of path B may be higher than path A since more NO is released in nimustine and we do not find the oxadiazolium cation as found in 1a. However, in BCNU the ESI-MS data shows evidence of the aziridinium cation (Scheme S4†) which was not found in 1a or nimustine. Hence, for BCNU path C is feasible. It should be noted that the path C cannot be totally excluded for 1a or nimustine since the stability of the aziridinium formed under the conditions of ESI-MS would dictate its detection. The results bring more clarity to why these N-(2-chloroethyl)-N-nitrosoureas are active against different type of cancer instead of all of them being selective to one particular type of cancer. One strong reason could be that they have different mechanistic pathway.
The amount of NO released per mole of compound would influence the cytotoxicity. 1a and 2a shows good cytotoxicity under the in vitro testing conditions although the NO release rate is higher for nimustine. The reason may be that the NO is released before the compound can enter the cell making it less active. The clinically approved BCNU as per literature has a half life of 15–20 min in plasma66 but 2a has a half-life of 39 min in buffer and shows better anticancer activity than BCNU on MCF-7 and A549. Apart from the compounds being more effective in the probed cell lines compared to BCNU and nimustine, the most important factor here is the NO release by the two aromatic N-(2-chloroethyl)-N-nitrosoureas and nimustine at physiologically relevant pH 6.0–7.4. The results of NO release studies confirm that nitric oxide release is happening at physiological pH and hence may influence the mechanism of action for the aromatic N-(2-chloroethyl)-N-nitrosoureas similar to that highlighted for aliphatic acyclic N-(2-chloroethyl)-N-nitrosoureas by the earlier work28,30 of Lown et al. We found that faster NO release, may not lead to better activity. The compound may be deactivated before it can reach the target if the NO release is faster. It should be borne in mind that NO release, is not the sole pathway of action for N-(2-chloroethyl)-N-nitrosoureas and our results also support the same. However, NO release is happening in all the compounds. More work has to be done, keeping the NO release pathway in mind so that we can gain more insight about the correlation of NO release rate and cytotoxicity.
Experimental
Materials and methods
All chemicals and solvents were purchased from commercial sources. Solvents were distilled and dried prior to use. UV-visible measurements were done using Perkin Elmer lambda 35 spectrophotometer. FT-IR spectra were recorded using Perkin-Elmer 120-000A in KBr pellets. Fluorescence measurements were carried out in Horiba Jobin Yvon Fluorolog instrument. The excitation source used here is a steady state 450 W Xe lamp. 1H & 13C NMR spectra were measured using Bruker Avance III 500 MHz spectrometer at room temperature. The chemical shifts are reported in parts per million (ppm). The proton decoupled 13C NMR spectra are reported. Elemental analyses were performed on a PerkinElmerEA 2400 CHNS series elemental analyzer. Electro-spray ionization mass spectra were recorded using micromass Q-Tof microTM, Waters by +ve mode electrospray ionization. The synthetic yields reported are of isolated analytically pure compounds.Syntheses
X-ray crystallography
Single crystal of 1a was mounted using loops on the goniometer head of a SuperNova, Dual, Cu at zero, EOS diffractometer. The crystal was kept at 100.00(10) K temperature during data collection. Using Olex2,67 the structure was solved with the Superflip68 structure solution program using Charge Flipping and refined with the ShelXL69 refinement package using Least Squares minimisation.Aqueous stability studies
The determination of the half life of the nitrosoureas was carried out by monitoring the decrease of absorbance in UV-vis spectrophotometer.32 5 μL (1 × 10−2 M) of acetonitrile stock solution of nitrosoureas were taken in 995 μL phosphate buffer (50 mM, pH = 7.4). Absorbances were recorded at 5 min time interval at 298 K. The aqueous decomposition rate constant were determined by plotting ln(Abs) vs. time data where from, the slope, rate of aqueous decomposition (kD) and corresponding half life (t1/2) were determined.Nitric oxide (NO) release studies
Headspace gas (NO) detection by Griess reagent. The NO release experiments were carried out using the headspace gas generated in the sealed vial of the DMSO–buffer solution (7
:3) of 1a, 2a and nimustine hydrochloride. The overhead gas were syringed out in a gas tight syringe (2 × 2 mL) and purged into a solution containing 200 μL freshly prepared Griess reagent (0.1% sulfanilide, 1% N-naphthylethylenediamine dihydrochloride and 1% phosphoric acid) and 600 μL 10 mM phosphate buffer (pH = 7.0). The increase in absorbance at 540 nm was measured with time. However, the reaction of NO with Griess reagent is slower and hence some amount of NO escapes the detection.
Solution phase Griess test. The NO release from 1a and 2a was confirmed by the estimation of nitrite using Griess reagent. To 600 μL of 10 mM phosphate buffer (pH = 7.0), 195 μL freshly prepared Griess reagent (0.1% sulfanilide, 1% N-naphthylethylenediamine dihydrochloride and 1% phosphoric acid) was added followed by the addition of compound solution (5 μL) of 1a and 2a in DMSO (1 × 10−2 M). The estimation of nitric oxide or nitrite ion was measured by observing the absorbance at 540 nm which continuously increased over the time period of the experiment. The data were recorded every 6 min time interval. Standard calibration curve was derived from 2–60 μM sodium nitrite standard solutions.
Fluorescence spectroscopy. Fluorescence of 50 μM 1,2-diaminoanthraquinone (DAA) solution in DMSO was recorded using λex = 490 nm. 8 μL of 5 mM DMSO solution of 1a (final conc. 50 μM) in 50 μM DAA solution was added and spectra recorded using λex = 490 nm. Same method was followed for nimustine hydrochloride. To detect nitric oxide in the headspace gas evolved from 1a, a 20 mM DMSO solution of 1a was prepared in a sealed vial and 20 mL headspace gas was syringed out in a gas tight syringe and purged into the 50 μM aerated DAA solution and after 10 min the spectra was recorded using λex = 490 nm.
pH and conductivity study. The NO release would lead to change in pH due to formation of nitrous/nitric acid hence the change in pH was monitored using an aqueous solution containing 20% DMSO and 1a. The pH decrease over time was recorded using a Mettler Toledo SevenEasy pH meter (Fig. S15†).
The increase in conductivity due to formation of acid by the released NO could also provide indication of NO generation. Hence, using the head space gas of a Teflon capped sealed vial containing 1a in an aqueous solution with 20% DMSO the conductivity was measured using a Mettler Toledo SevenGo conductivity meter (Fig. S16†).
ESI-MS studies
ESI-MS were recorded using micromass Q-Tof microTM, Waters and Bruker maXis Impact (ESI-QTOF) in +ve mode. The samples were prepared in acetonitrile or methanol solution and for mechanistic pathways determination the samples were prepared in acetonitrile–water mixture (2:1) and immediately recorded. We even tried other aqueous mixtures viz. methanol–water (2:1 v/v), DMSO–water (0.5:9.5 v/v). All of these solvent mixtures gave the same m/z pattern but the intensities were better with acetonitrile–water (2:1 v/v).
Cell lines and culture condition
Human breast adenocarcinoma cell line (MCF-7), human lung carcinoma cell line (A549), human embryonic kidney cell (HEK293T) and normal mouse embryonic fibroblast cell line (NIH3T3) were kindly provided by Department of Biological Sciences, IISER-Kolkata, India. The cell lines were maintained in the logarithmic phase at 37 °C in a 5% carbon dioxide atmosphere using a culture media containing DMEM, 10% foetal bovine serum (GIBCO), antibiotics (100 units mL−1 penicillin and 100 μg mL−1 streptomycin). In hypoxic condition the oxygen percentage was maintained at 1.5%.Cell viability assay
The cytotoxicity of compounds were evaluated on MCF-7, A549, HEK293T and NIH3T3 cell lines by MTT assay.70 Briefly, 6 × 103 cells per well, were seeded in 96-well plates in DMEM (200 μL) and then incubated at 37 °C in a 5% carbon dioxide atmosphere. After 48 h, the media was removed and replaced with a fresh one. Compounds to be studied were added at appropriate concentrations. Each concentration was tested in triplicates in the wells. The compounds to be added were first solubilised in media or PBS containing DMSO (when needed) such that the concentration of DMSO in well should not exceed 0.2%. BCNU and nimustine were solubilised in acidic ethanol followed by dilution with water.For hypoxic condition the O2% of the CO2 incubator (ESCO cell culture CO2 incubator, model: CCL-170T-8-UV) was maintained at 1.5%. Even for incubation under hypoxia 48 h period was used. The drug additions were all done in normoxic condition and the 96 well plates were then placed in incubator. It took ca. 30 min for the incubator to reach oxygen percentage at 1.5%.
After 48 h the drug containing media was removed and fresh media was added to each well and successively treated with 20 μL of a 1 mg mL−1 MTT in 1× PBS (pH = 7.2). After 3 h of incubation, media was removed and 200 μL of DMSO were added to each well. The inhibition of cell growth induced by the tested complexes was detected by measuring the absorbance of each well at 515 nm (ref. 71 and 72) using a BIOTEK ELx800 plate reader. The obtained data were plotted and fitted using GraphPad Prism 5® Ver 5.03 and shown in Fig. S5–S10.†
Statistical analysis
All the IC50 data are given are mean ± standard deviation. The results are mean of three independent experiments carried out in each cell line where, in each experiment each concentration was assayed in triplicate. The statistical analyses were performed using Graph pad prism® software 5.0 with student's t-test.Cell cycle arrest
A549 cells (1 × 106 per plate) were grown in 100 mm dia petri dish suspended in 12 mL DMEM media at previously mentioned culturing condition. After 48 h, media was removed followed by addition of fresh media. Appropriate concentration of compound solution of 2a were added and incubated at same condition as above. After 24 h drug exposure, cells were harvested by trypsinization, centrifuged and washed twice with cold 1× PBS buffer (pH = 7.2). Cells were again resuspended in 100 μL cold 1× PBS buffer and fixed with 70% aqueous ethanol for overnight at 4 °C. DNA staining was done by resuspending the cell pellets in 1× PBS solution containing PI (55 μg mL−1) and RNase A (100 μg mL−1) solution. Cell suspensions were gently mixed and incubated at 37 °C for half an hour. Then samples were analyzed in a BD Biosciences FACSCalibur flow cytometer.Fluorescence microscopy
A549 cells, 12 × 103 cells per well, were seeded in a 6-well plate with 3 mL of DMEM media and incubated for 48 h in afore-mentioned culture condition. After incubation, fresh media was added and drug solutions were added. After 24 h of drug exposure, cells were fixed with 4% paraformaldehyde solution in 1× PBS (pH 7.2). Then the staining was performed with 300 nM of DAPI solution. After several times washing with cold 1× PBS (pH 7.2), optical microscopy images of A549 cells were acquired using OLYMPUS IX 81 epifluorescence inverted microscope at 60× magnification. Both DIC and fluorescence microscopy images were taken and processed using OLYMPUS Cell P software.Conclusions
The N-(2-chloroethyl)-N-nitrosoureas derived from aromatic amines or nimustine hydrochloride may involve release of NO in their mechanistic pathway of action for anticancer activity. The results suggest that both the compound 1a and 2a are most active against A549 (IC50 = 38 ± 1 μM and 41 ± 5 μM) and are equally good even in hypoxia which is not always so common.73,74 The aqueous decomposition rate does not necessarily correlate with the in vitro cytotoxicity data. 2a arrests the cell cycle of A549 in G2/M-phase. Nimustine which was found to be least toxic showed a higher NO release rate even at physiological pH as per the overhead gas studies. Hence, it appears that if NO release occurs faster then the compound may become less active but if the NO release is slow the compounds may be more active. In addition the results suggest that the mechanistic pathways of action of N-(2-chloroethyl)-N-nitrosoureas involves multiple dissociative pathways and NO release is not the sole pathway of action as suggested by earlier work in this area. Nevertheless our work shows that NO release should be taken into account due to its significant occurrence at physiological pH. The above work not only highlights a new possible mechanistic pathway of action for N-(2-chloroethyl)-N-nitrosoureas but also shows that aromatic N-(2-chloroethyl)-N-nitrosoureas may be potent anticancer agents based on their in vitro studies in comparison to BCNU and nimustine.Acknowledgements
We sincerely acknowledge DST for the funding (Vide Project no. SR/S1/IC-36/2010). We thank Dr Jayasri Das Sarma, Dr Tapas K Sengupta and Dr Partho Sarothi Ray, Department of Biological Sciences, IISER Kolkata, India for kind donation of the cell lines. We are also thankful to IISER Kolkata for the financial and infrastructural support including NMR, FACS and ESI-MS, single crystal X-ray diffraction facility. AS sincerely acknowledges IISER Kolkata for research fellowship. SK and KP thank UGC and SB thanks CSIR respectively for research fellowship.References
- C. T. Gnewuch and G. Sosnovsky, Chem. Rev., 1997, 97, 829–1013 CrossRef CAS PubMed.
- S. R. Rajski and R. M. Williams, Chem. Rev., 1998, 98, 2723–2795 CrossRef CAS PubMed.
- V. A. Levin and C. B. Wilson, Cancer Treat. Rep., 1976, 60, 719–724 CAS.
- F.-X. Chen, W. J. Bodell, G. Liang and B. Gold, Chem. Res. Toxicol., 1996, 9, 208–214 CrossRef CAS PubMed.
- G. P. Wheeler, B. J. Bowdon, J. A. Grimsley and H. H. Lloyd, Cancer Res., 1974, 34, 194–200 CAS.
- G. P. Wheeler, B. J. Bowdon and R. F. Struck, Cancer Res., 1975, 35, 2974–2984 CAS.
- S. K. Carter, F. M. Schabel Jr, L. Broder and T. P. Johnston, Adv. Cancer Res., 1972, 16, 273–332 CrossRef CAS.
- I. Petak, R. Mihalik, P. I. Bauer, H. Suli-Vargha, A. Sebestyen and L. Kopper, Cancer Res., 1998, 58, 614–618 CAS.
- H.-S. Gwak, S.-M. Youn, A.-H. Kwon, H. Lee Seung, H. Kim Jong and H. Rhee Chang, J. Neuro-Oncol., 2005, 75, 173–180 CrossRef CAS PubMed.
- S. Scoccianti, B. Detti, A. Sardaro, A. Iannalfi, I. Meattini, B. G. Leonulli, S. Borghesi, F. Martinelli, L. Bordi, F. Ammannati and G. Biti, Anti-Cancer Drugs, 2008, 19, 613–620 CrossRef CAS PubMed.
- R. Addeo, M. Caraglia, M. S. Santi, L. Montella, A. Abbruzzese, C. Parlato, B. Vincenzi, M. Carraturo, V. Faiola, M. Genovese, G. Cennamo and S. Prete, J. Neuro-Oncol., 2011, 102, 417–424 CrossRef CAS PubMed.
- M. Santoni, S. Scoccianti, I. Lolli, M. G. Fabrini, G. Silvano, B. Detti, F. Perrone, G. Savio, R. Iacovelli, L. Burattini, R. Berardi and S. Cascinu, J. Neuro-Oncol., 2013, 113, 397–401 CrossRef CAS PubMed.
- S. Shibui, Y. Narita, J. Mizusawa, T. Beppu, K. Ogasawara, Y. Sawamura, H. Kobayashi, R. Nishikawa, K. Mishima, Y. Muragaki, T. Maruyama, J. Kuratsu, H. Nakamura, M. Kochi, Y. Minamida, T. Yamaki, T. Kumabe, T. Tominaga, T. Kayama, K. Sakurada, M. Nagane, K. Kobayashi, H. Nakamura, T. Ito, T. Yazaki, H. Sasaki, K. Tanaka, H. Takahashi, A. Asai, T. Todo, T. Wakabayashi, J. Takahashi, S. Takano, T. Fujimaki, M. Sumi, Y. Miyakita, Y. Nakazato, A. Sato, H. Fukuda and K. Nomura, Cancer Chemother. Pharmacol., 2013, 71, 511–521 CrossRef CAS PubMed.
- J. P. G. Brakenhoff, J. N. M. Commandeur, L. W. Wormhoudt, E. J. Groot and N. P. E. Vermeulen, Carcinogenesis, 1996, 17, 715–724 CrossRef CAS PubMed.
- T. Moritz, W. Mackay, B. J. Glassner, D. A. Williams and L. Samson, Cancer Res., 1995, 55, 2608–2614 CAS.
- C. C. Bailey, H. B. Marsden and P. H. Jones, Fatal pulmonary fibrosis following 1,3-bis(2-chloroethyl)-1-nitrosourea (BCNU) therapy, United States FIELD Citation, 1978 Search PubMed.
- T. P. Johnston, G. S. McCaleb and J. A. Montgomery, J. Med. Chem., 1963, 6, 669–681 CrossRef CAS.
- F. M. Schabel Jr, T. P. Johnston, G. S. McCaleb, J. A. Montgomery, W. R. Laster and H. E. Skipper, Cancer Res., 1963, 23, 725–733 CAS.
- T. P. Johnston, G. S. McCaler, P. S. Opliger, W. R. Laster and J. A. Montgomery, J. Med. Chem., 1971, 14, 600–614 CrossRef CAS.
- T. P. Johnston, G. S. McCaleb, S. D. Clayton, J. L. Frye, C. A. Krauth and J. A. Montgomery, J. Med. Chem., 1977, 20, 279–290 CrossRef CAS.
- A. M. Crider, T. M. Kolczynski and K. M. Yates, J. Med. Chem., 1980, 23, 324–326 CrossRef CAS.
- A. M. Crider, R. Lamey, H. G. Floss, J. M. Cassady and W. J. Bradner, J. Med. Chem., 1980, 23, 848–851 CrossRef CAS.
- M. Iwasaki, M. Ueno, K. Ninomiya, J. Sekine, Y. Nagamatsu and G. Kimura, J. Med. Chem., 1976, 19, 918–923 CrossRef CAS.
- T.-S. Lin, P. H. Fischer, G. T. Shiau and W. H. Prusoff, J. Med. Chem., 1978, 21, 130–133 CrossRef CAS.
- M. R. Berger, J. Floride, D. Schmaehl, J. Schreiber and G. Eisenbrand, Eur. J. Cancer Clin. Oncol., 1986, 22, 1179–1191 CrossRef CAS.
- K. A. Kennedy, B. A. Teicher, S. Rockwell and A. C. Sartorelli, Biochem. Pharmacol., 1980, 29, 1–8 CrossRef CAS.
- W. J. Zeller, K. Ehresmann and G. Eisenbrand, J. Cancer Res. Clin. Oncol., 1984, 108, 249–251 CrossRef CAS.
- J. W. Lown and S. M. S. Chauhan, J. Org. Chem., 1981, 46, 5309–5321 CrossRef CAS.
- J. W. Lown and S. M. S. Chauhan, J. Med. Chem., 1981, 24, 270–279 CrossRef CAS.
- J. W. Lown and S. M. S. Chauhan, J. Org. Chem., 1983, 48, 3901–3908 CrossRef CAS.
- G. Eisenbrand, S. Lauck-Birkel and W. C. Tang, Synthesis, 1996, 1247–1258 CAS.
- S. L. Aukerman, R. B. Brundrett, J. Hilton and P. E. Hartman, Cancer Res., 1983, 43, 175–181 CAS.
- J. A. Montgomery, R. James, G. S. McCaleb, M. C. Kirk and T. P. Johnston, J. Med. Chem., 1975, 18, 568–571 CrossRef CAS.
- A. Naghipur, M. G. Ikonomou, P. Kebarle and J. W. Lown, J. Am. Chem. Soc., 1990, 112, 3178–3187 CrossRef CAS.
- M. D. Bacolod, R. Fehdrau, S. P. Johnson, N. S. Bullock, D. D. Bigner, M. Colvin and H. S. Friedman, Cancer Chemother. Pharmacol., 2009, 63, 753–758 CrossRef CAS PubMed.
- J. K. Snyder and L. M. Stock, J. Org. Chem., 1980, 45, 1990–1999 CrossRef CAS.
- J. W. Lown and S. M. S. Chauhan, J. Org. Chem., 1982, 47, 851–856 CrossRef CAS.
- S. Amado, L. Garcia-Rio, J. R. Leis and A. Rios, J. Chem. Soc., Perkin Trans. 2, 1996, 2235–2239 RSC.
- D. R. Janero, Free Radicals Biol. Med., 2000, 28, 1495–1506 CrossRef CAS.
- T. Yamamoto and R. J. Bing, Proc. Soc. Exp. Biol. Med., 2000, 225, 200–206 CrossRef CAS.
- M. Feelisch, Naunyn-Schmiedeberg's Arch. Pharmacol., 1998, 358, 113–122 CrossRef CAS.
- P. G. Wang, M. Xian, X. Tang, X. Wu, Z. Wen, T. Cai and A. J. Janczuk, Chem. Rev., 2002, 102, 1091–1134 CrossRef CAS PubMed.
- R. M. J. Palmer, A. G. Ferrige and S. Moncada, Nature, 1987, 327, 524–526 CrossRef CAS PubMed.
- L. J. Ignarro, G. M. Buga, K. S. Wood, R. E. Byrns and G. Chaudhuri, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 9265–9269 CrossRef CAS.
- E. Culotta and D. E. Koshland Jr, Science, 1992, 258, 1862–1865 CAS.
- L. J. Ignarro, Angew. Chem., Int. Ed., 1999, 38, 1882–1892 CrossRef CAS.
- F. Murad, Angew. Chem., Int. Ed., 1999, 38, 1856–1868 CrossRef CAS.
- R. F. Furchgott, Angew. Chem., Int. Ed., 1999, 38, 1870–1880 CrossRef CAS.
- R. Scatena, P. Bottoni, A. Pontoglio and B. Giardina, Curr. Med. Chem., 2010, 17, 61–73 CrossRef CAS.
- E. Nava and T. F. Luscher, J. Hypertens., 1995, 13, S39–S48 CrossRef CAS PubMed.
- P. Kubes, M. Suzuki and D. N. Granger, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 4651–4655 CrossRef CAS.
- P. N. Coneski and M. H. Schoenfisch, Chem. Soc. Rev., 2012, 41, 3753–3758 RSC.
- S. Mocellin, V. Bronte and D. Nitti, Med. Res. Rev., 2007, 27, 317–352 CrossRef CAS PubMed.
- D. A. Riccio and M. H. Schoenfisch, Chem. Soc. Rev., 2012, 41, 3731–3741 RSC.
- L. Gao and J. L. Williams, Int. J. Oncol., 2012, 41, 325–330 CAS.
- T. P. Johnston, G. S. McCaleb, P. S. Opliger and J. A. Montgomery, J. Med. Chem., 1966, 9, 892–910 CrossRef CAS.
- H. W. Smith, A. Camerman and N. Camerman, J. Med. Chem., 1978, 21, 468–471 CrossRef CAS.
- S. L. Bekoe, D. Urmann, A. Lakatos, C. Glaubitz and M. U. Schmidt, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2012, 68, o144–o148 CAS.
- O. Tredan, C. M. Galmarini, K. Patel and I. F. Tannock, J. Natl. Cancer Inst., 2007, 99, 1441–1454 CrossRef CAS PubMed.
- N. Raghunand, X. He, R. van Sluis, B. Mahoney, B. Baggett, C. W. Taylor, G. Paine-Murrieta, D. Roe, Z. M. Bhujwalla and R. J. Gillies, Br. J. Cancer, 1999, 80, 1005–1011 CrossRef CAS PubMed.
- K. Ghosh, S. Kumar, R. Kumar and U. P. Singh, Eur. J. Inorg. Chem., 2012, 2012, 929–938 CrossRef CAS.
- H. Kojima and T. Nagano, Adv. Mater., 2000, 12, 763–765 CrossRef CAS.
- T. Nagano, Luminescence, 1999, 14, 283–290 CrossRef CAS.
- T. Nagano and T. Yoshimura, Chem. Rev., 2002, 102, 1235–1269 CrossRef CAS PubMed.
- M. J. Marin, P. Thomas, V. Fabregat, S. V. Luis, D. A. Russell and F. Galindo, ChemBioChem, 2011, 12, 2471–2477 CrossRef CAS PubMed.
- T. L. Loo, R. L. Dion, R. L. Dixon and D. P. Rall, J. Pharm. Sci., 1966, 55, 492–497 CrossRef CAS.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- L. Palatinus and G. Chapuis, J. Appl. Crystallogr., 2007, 40, 786–790 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS.
- B. L. Lokeshwar, E. Escatel and B. Zhu, Curr. Med. Chem., 2001, 8, 271–279 CrossRef CAS.
- M. C. Alley, D. A. Scudiero, A. Monks, M. L. Hursey, M. J. Czerwinski, D. L. Fine, B. J. Abbott, J. G. Mayo, R. H. Shoemaker and M. R. Boyd, Cancer Res., 1988, 48, 589–601 CAS.
- S. Koch, F. Mayer, F. Honecker, M. Schittenhelm and C. Bokemeyer, Br. J. Cancer, 2003, 89, 2133–2139 CrossRef CAS PubMed.
- B. A. Teicher, J. S. Lazo and A. C. Sartorelli, Cancer Res., 1981, 41, 73–81 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Figures showing aqueous decomposition of compound 1–2a and nimustine hydrochloride in buffer (pH −7.4, 6.0), fitting plots for the determination of compounds 1a and 2a, cell cycle analysis of 2a and nitric oxide release study of 1a, 2a and nimustine hydrochloride, 1H and 13C NMR spectra of 1, 2, 1a and 2a. CCDC 1019082. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ra11137k |
| ‡ The authors pay equal contributions to this work. |
| This journal is © The Royal Society of Chemistry 2015 |