
Enhanced electrochemical performance of Li2NiTiO4 with micro-structural rearrangement via urea treatment
Jingjing Wuab,
Haibo Wangab,
Jinbing Quanab,
Zheng Maab and
Decheng Li*ab
aCollege of Physics, Optoelectronics and Energy & Collaborative Innovation Center of Suzhou Nano Science and Technology, Soochow University, Suzhou 215006, China. E-mail: lidecheng@suda.deu.cn; Fax: +86-512-67261575; Tel: +86-512-67261337
bKey Laboratory of Lithium Ion Battery Materials of Jiangsu Province, Institute of Chemical Power Sources, Soochow University, Suzhou 215006, China
First published on 26th November 2014
Abstract
Nanosized Li2NiTiO4 (LNT) with a disordered rock salt structure has been prepared by the sol–gel method. To improve the electrochemical performance, LNT has been mixed with urea and sintered in N2 atmosphere. The effects of different sintering temperatures in N2 on the micro-structural and electrochemical performances have been investigated by X-ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS), N2 adsorption–desorption isotherm, transmission electron microscopy (TEM) and charge–discharge test. The electrochemical results show that Li2NiTiO4 with urea treatment at 500 °C (U-LNT-500) has the best rate capability, the highest discharge capacity and the best cycle ability, indicating that the electrochemical performances of LNT could be remarkably improved by this treatment. It is interesting to note that the discharge capacity of U-LNT-500 at ambient temperature is almost as high as the discharge capacity of LNT at 50 °C. It is believed that the micro-structural changes, such as the increase of the specific surface area and the volume of pores as well as the local ordered structure in terms of atomic arrangement after urea treatment should account for the enhancement of LNT electrochemical performances.
1. Introduction
Global warming and air pollution due to the combustion of fossil fuels have sparked the need of replacement of internal combustion engines with electric motors, such as hybrid electric vehicles (HEVs), plug-in hybrid electric vehicles (PHEVs) and electric vehicles (EVs). Lithium-ion batteries (LIB) as a clean power source can make it become reality by powering these electric motors, because LIB delivers the stored electrochemical energy as electrical energy with high conversion efficiency and without any gaseous emission. Also owing to the high energy density, LIB has been the power source for the portable electronic products like cellular phone, digital cameras, and laptop and so on. Therefore, the LIB developed for the high-energy transportation sector has been received intensive attention. Nevertheless, there are many issues like lower cost, safety, lifetime, and especially the power and energy densities needed to be concerned. To meet these requirements, the direct strategy is to develop new electrode materials, although optimizing LIB structures can provide limited improvement.1–3 LiCoO2 has been the dominant cathode material for LIB over the past two decades because of its stable electrochemical cycling performance. However, the high cost, toxicity, and safety problem prevent its use for transportation.4 LiMn1.5Ni0.5O4 is considered to be an alternative cathode material for LIB. The advantages of this compound are its high operating voltage of 4.7 V and a specific capacity delivered close to its theoretical value (148 mA h g−1). The both characters make LiMn1.5Ni0.5O4 a promising cathode material for high-energy applications. One factor hindering its utilizing is the decomposition of organic electrolytes when the battery is charged to higher voltage of >4.5 V,5 and the other is a severe capacity fade at elevated temperature (60 °C).6 Lithium-rich Li[Li, Mn, Ni, Co]O2, another cathode material suitable for the high-energy applications, can achieve the discharge capacity of 250–300 mA h g−1 with an operating voltage window of 2.0–4.8 V. Unfortunately, it suffers from several issues such as voltage fade, impedance rise, the huge irreversible capacity loss of 40–100 mA h g−1 in the first cycle and the poor rate capability.7,8 In recent years, the group of the general formula Li2MTiO4 (M = Mn, Fe, Co, Ni) cathode materials has been proposed as a potential commercial material, because its capacity can achieve as high as 290 mA h g−1 if the lithium ions can completely extracted in charge–discharge process.9–13 In addition, Li2MTiO4 has other appealing features, such as high safety, environmental friendliness and low cost. Nevertheless, Li2MTiO4 has a cation disordered structure with the space group Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m. In the disordered structure, metal ions are randomly distributed in octahedral 4a positions, which results in the lack of lithium ions diffusion pathways and poor electrochemical performance. Besides these, the poor electronic conductivity is another drawback of Li2MTiO4.
m. In the disordered structure, metal ions are randomly distributed in octahedral 4a positions, which results in the lack of lithium ions diffusion pathways and poor electrochemical performance. Besides these, the poor electronic conductivity is another drawback of Li2MTiO4.
In the Li2MTiO4 (M = Mn, Fe, Co, Ni) family, Li2NiTiO4 has attracted lots of researches because of its many advantages such as the ease synthesis process and its high purity. Additionally, the most prominent feature is that it has two structures. One is disordered rock salt structure and the other is ordered monoclinic structure. This ordered monoclinic structure with the space group C12/c1 consists of lithium-poor [Li1/3(Ni, Ti)2/3] and lithium-rich [Li2/3(Ni, Ti)1/3] cation layers alternate in the (111)cubic planes of rock salt anion array. It has been synthesized through a low temperature method by Sebastian et al.9 Unfortunately, this synthesis process needed as long as 7 days. Later, S. R. S. Prabaharan et al. used the citric acid-assisted low temperature solution method to get a Li2NiTiO4 nanoparticle with rock salt structure.12 The first charge capacity could achieve to 181 mA h g−1 at 0.5 C between 2.5 V and 4.8 V. However, the capacity fading of this material almost arrived to 50% in the first cycle. Kuezma et al. studied the electrochemical performance of this disordered Li2NiTiO4 with LiBOB electrolyte in the potential range of 2.5–4.8 V at 0.5 C.13 The results showed that the fortieth discharge capacity retention was only 36% at 25 °C and also showed the rapid capacity decay in the process of continuous cycle at 60 °C. Yusuke Kawano et al. used the Si4+ to substitute Ti4+ and got the Li2NiSi0.1Ti0.9O4.14 Although the initial charge and discharge were increased, the first discharge–charge efficiency was declined instead and the improvement of cycle ability was also restricted. One of the reasons could be that the highly charged Ni(IV) ions hindered the movement of ions and electrons during charge process, resulting in the Li-ions irreversible rearrangement (migrating from octahedral 4a to tetrahedral 8c sites) and the depravation of electronic conductivity.10 In the case of the Li4Ti5O12 which is a promising anode material and has the same problem, it was reported that urea (a safe and inexpensive material) treatment could effectively improve its electronic conductivity, which has been ascribed to the form of a thin and dense TiN layer on the particle surface.15
In this study, we adopted urea treating the disordered Li2NiTiO4 and studied the effect of urea treatment on the electrochemical performance of Li2NiTiO4. Results show that capacity and rate capability are both improved remarkably.
2. Experimental section
2.1 Synthesis process
The compound (Li2NiTiO4) was synthesized using a low temperature citric-acid assisted sol–gel method. Stoichiometric amounts of LiNO3, Ni(NO3)2·6H2O and Ti(OC3H7)4 were separately dissolved in absolute ethanol and were mixed together under vigorous agitation. Then, the citric acid with weight ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to total weight of metal precursors was slowly added into above mixed solution, followed by stirring constantly for 30 min at 80 °C. Subsequently the temperature was increased to 120 °C to evaporate excess solvent for several hours until a green transparent gel was formed. The gel was dried in an oven at 80 °C for 24 h. The dried gel got was pre-sintered at 300 °C in air for 4 h to decompose the organic constituents. After grounded to form powders, the precursor was sintered at 550 °C for 12 h in a muffle furnace to obtain Li2NiTiO4 (denoted as LNT).
:1 to total weight of metal precursors was slowly added into above mixed solution, followed by stirring constantly for 30 min at 80 °C. Subsequently the temperature was increased to 120 °C to evaporate excess solvent for several hours until a green transparent gel was formed. The gel was dried in an oven at 80 °C for 24 h. The dried gel got was pre-sintered at 300 °C in air for 4 h to decompose the organic constituents. After grounded to form powders, the precursor was sintered at 550 °C for 12 h in a muffle furnace to obtain Li2NiTiO4 (denoted as LNT).
2.2 Urea treatment
The as-prepared Li2NiTiO4 powders were mixed uniformly with a 6 wt% ratio of urea in an aqueous solution under constantly stirring. After the thorough evaporation of water at 70 °C, the mixture was dried in the oven at 60 °C overnight and then calcined for 30 min under N2 atmosphere at different temperatures of 500 °C and 600 °C. The obtained samples were denoted as U-LNT-500 and U-LNT-600, correspondingly.2.3 Electrode fabrication
The coin cell (CR2032) was used to test the electrochemical performance. The cathode electrodes were prepared by mixing 80 wt% active materials, 10 wt% Super-p and 10 wt% polyvinylidene fluoride (PVDF) binders homogeneously mixed in N-methyl pyrrolidinone (NMP) to form slurry. Then the slurry was spread on aluminum foil, and was dried at 110 °C for 12 h in the vacuum oven to remove any residual solvent and possible adsorbed water. The mass loading in the electrode was controlled at about 4.2 mg cm−2. The cell was consisted of as-prepared working electrode, lithium metal as the counter electrode, Celgard 2400 porous polypropylene film as separator and 0.2 mL electrolyte. The electrolyte was 1 M LiPF6 in a mixture of ethylene carbonate–diethyl carbonate (EC–DEC, 1:1 by volume).
2.4 Characterization
XRD analysis was carried out using a X'Pert-ProMPD (Holand) D/max-A X-ray diffractometer with Cu Kα radiation (λ = 0.154178 nm) between 10° and 80° at a scan rate of 0.04°. TEM and HRTEM were taken on a FEI-Tecnai G 20 (200 kV) to obtain the particulate morphology and structure. XPS measurements were carried with an ESCALAB 250Xi system with Al Kα radiation as the exciting source. The binding energies were calibrated by referencing the universal carbon contamination peak at 284.8 eV. N2 adsorption–desorption isotherms were carried out at 77 K, using an ASAP2050 (Micromeritics Industrument Corp.) surface area & porosity Analyzer. The specific surface area and the pore size distribution were calculated by the Brunauer–Emmett–Teller (BET) method and the Barret–Joyner–Halenda (BJH) method, respectively. The discharge–charge experiments were tested over a voltage range of 1.5–4.8 V (vs. Li+/Li) at different rates on a LAND-CT2001A battery test system (Jinnuo Wuhan Co., Ltd., P. R. China).3. Results and discussion
Fig. 1 shows the XRD patterns for samples before and after urea treatment. The diffraction peaks of LNT can be indexed in the typical rock salt structure with the space group Fmm, which was well consistent with previous report.12 After urea treatment, no other diffraction peaks can be detected, suggesting that no impurity was formed during urea treatment process. Compared with LNT, the diffraction peaks of U-LNT-500 slightly move to lower angles, while U-LNT-600 showed very little shifts to higher angles, implying the lattice expansion for the sample treated at 500 °C, and lattice shrinks for the sample treated at 600 °C. The lattice parameters for the three samples were calculated by the least squares method and values are listed in Table 1. The lattice constants are 4.1245, 4.1342 and 4.1204 Å for LNT, U-LNT-500, and U-LNT-600, respectively. Meanwhile, the cell volumes are 70.17, 70.66 and 69.95 Å3, correspondingly.
 | ||
| Fig. 1 X-ray diffraction patterns of LNT, U-LNT-500, and U-LNT-600. | ||
| Sample | a (Å) | V (Å3) |
|---|---|---|
| LNT | 4.1245 | 70.17 |
| U-LNT-500 | 4.1342 | 70.66 |
| U-LNT-600 | 4.1204 | 69.95 |
XPS study was carried out for the sake of confirming the oxidation state of the transition metal species before and after urea treatment. Fig. 2 shows the Ni, Ti and O XPS core level spectra for the three samples. The Ni2p spectra in Fig. 2a are characterized by two main lines Ni2p3/2 and Ni2p1/2 and two satellites S1 and S2. The appearance of satellite peaks could be ascribed to the multiple splitting which corresponds to the excited state in the energy bands.16 For the sample LNT, the peak at the binding energy (BE) values of 855.7 eV can be assigned to Ni2+ and the peak at 859.6 eV indicates the presence of Ni3+.17–20 After the LNT was mixed with urea and treated in N2, for the sample of U-LNT-500, only one peak corresponding to Ni2+2p3/2 at 855.6 eV was discovered, and no matched Ni3+2p3/2 peak was detected. Whereas two peaks at 855.3 eV and 858.3 eV were observed in the sample of U-LNT-600, which belong to Ni2+ and Ni3+, respectively. In Fig. 2b, all of the Ti2p peaks were composed of two bands. As can be observed from LNT, two peaks at 458.2 and 464.1 eV are generally known as the feature of Ti+4.21–24 Compared with LNT, the Ti2p3/2 and Ti2p1/2 peaks have shifts around 0.2 eV to lower BE values for the samples treated by urea. These small shifts could be negligible and no other peaks were found, implying no Ti4+ was reduced to Ti3+ by urea treatment. In previous reports about Li4Ti5O12 material, the TiN coating layer was introduced to improve the rate capability of Li4Ti5O12 because of its high electron conductivity.15 The formation of the TiN layer suggests that Ti4+ will be reduced to Ti3+. In this study, no Ti3+ was detected, indicating that no TiN layer was produced on the surface of Li2NiTiO4. In the O1s spectra (Fig. 2c), only one feature peak around 529.7 eV is really ascertained in the three samples, corresponding to the O2− ion in the metal oxide framework. Therefore, the XPS analysis shows that the urea treatment only has an effect on nickel ions. The reciprocal transformation of Ni2+ and Ni3+ coincides with lattice parameters changes in XRD. The lattice expansion for U-LNT-500 originates from the partial reduction of Ni3+ (0.60 Å) in the LNT to Ni2+ (0.69 Å) during urea treating process.
 | ||
| Fig. 2 XPS spectra for Ni2p (a), Ti2p (b), O1s (c) of LNT, U-LNT-500, and U-LNT-600. | ||
To clarify the electrochemical behaviours of the three samples before and after urea treatment, Fig. 3a compares the charge–discharge profiles of the three samples in the first cycle at 0.1 C. The three electrodes show similar charge–discharge profiles. All of the charge–discharge processes above 2.5 V are based on the redox species of Ni ions, and the discharge plateaus at around 2 V are corresponded to Ti ions.12,25,26 It is apparent that the charge–discharge capacities of LNT are lower than capacities of samples after urea treatment, in which U-LNT-500 shows more noticeable increment. As depicted in Fig. 3a, the first charge and discharge capacities of LNT are 130 and 95 mA h g−1, while those of U-LNT-500 are 168 and 130 mA h g−1, respectively. Besides, the voltage platform differences (ΔV) of LNT, U-LNT-500 and U-LNT-600 between charge and discharge curves are 0.5 V, 0.2 V and 0.3 V, respectively. Thus the ΔV is decreased after urea treatment and the ΔV of U-LNT-500 is the smallest, and this trend was kept at 2 C when the same cell was cycled to the fiftieth cycle with the charge–discharge rates from 0.1 C to 2 C, and each stage sustained for 10 cycles, which indicate that the inner resistance of the electrodes is reduced inside the material after urea treatment. These results suggest that urea treatment at 500 °C plays an important role in improving the charge–discharge capacities and reducing the inner resistance. The rate capacity properties were provided in Fig. 3c. As the charge–discharge rate increasing from 0.1 to 0.3, 0.5, 1, and 2 C, the discharge capacity of LNT drops remarkably from 100 to 81, 71, 57, and 37 mA h g−1, respectively, and the capacity retention was only 37% at 2 C (relative to 0.1 C), While the discharge capacity of U-LNT-500 decreases from 127 to 109, 99, 85, and 64 mA h g−1 separately, and the capacity retention can reach 50% at 2 C. For U-LNT-600, the discharge capacities at every stage and capacity retention rate (42%) are also improved, although not very noticeable. More importantly, for samples after treated by urea, when the discharge rate was finally returned back to 0.1 C, their capacities could nearly go back to the original values, while LNT could not, which proved the favourable structure reversibility of the electrode after urea treatment. The phenomenon discussed above suggested that the higher charge–discharge capacities, the enhanced rate capability and lower inner resistance can be easily archived by the urea treatment, especially when the treating temperature is 500 °C.
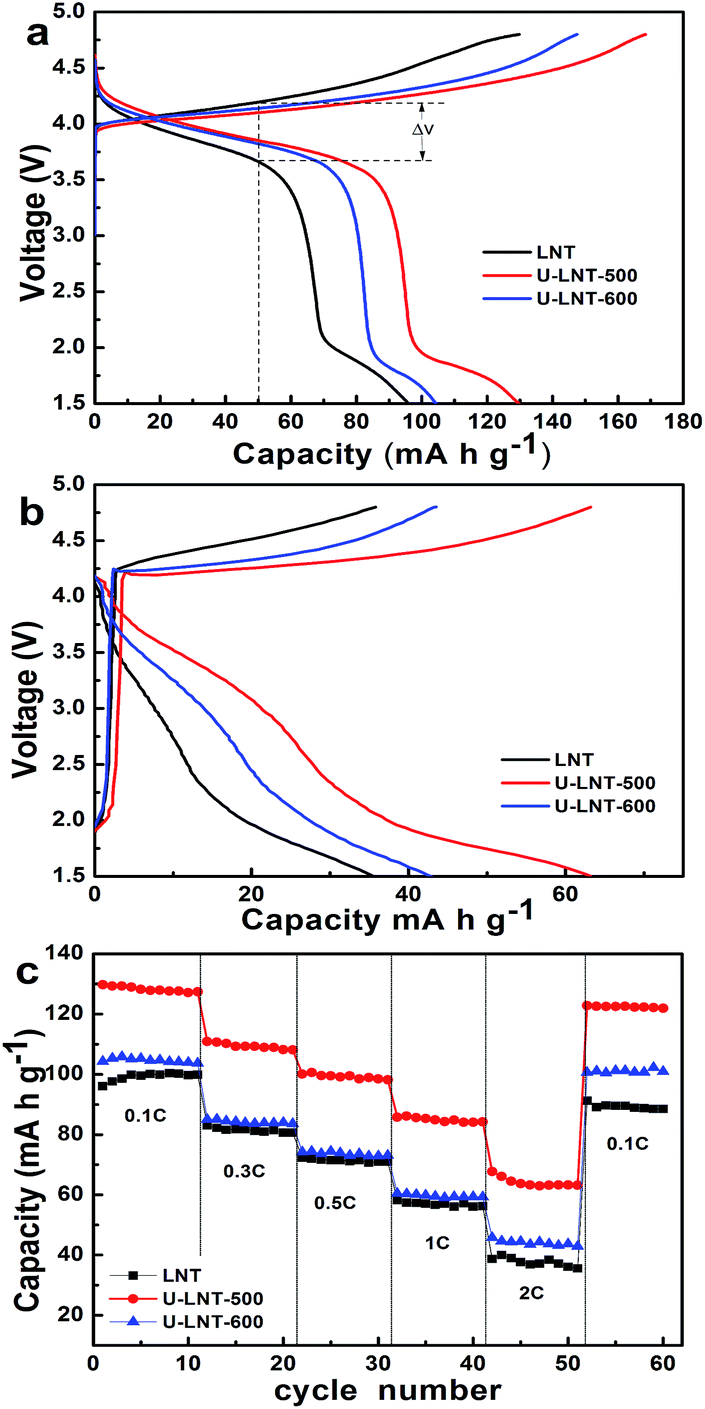 | ||
| Fig. 3 The charge–discharge curves of the three samples (a) at 0.1 C in the first cycle, and (b) at 2 C in the fiftieth cycle after charged–discharged from 0.1 C to 2 C; the rate performances (c) in the voltage range of 1.5–4.8 V. | ||
Fig. 4a presents the cycle performances for the three samples at the charge–discharge rate of 0.5 C at ambient temperature. Among the first seven cycles, the three samples exhibit a similar rule: capacity rising with the cycle numbers. This may be the reason of the activation of the materials. For LNT, The seventh and the fiftieth cycle capacities are 58 and 57 mA h g−1, respectively. This low discharge capacity can be ascribed to the disordered structure as anticipated, in which the lithium ions transport pathways are hindered because of the cations mixing. U-LNT-500 and U-LNT-600 exhibit the seventh cycle capacities of 96 and 69 mA h g−1 and the fiftieths of 92 and 67 mA h g−1, corresponding to the capacity retentions of 96% and 97%, respectively, which indicates that, after urea treatment, the great cycling ability was nearly maintained, and also the discharge capacity was greatly increased.
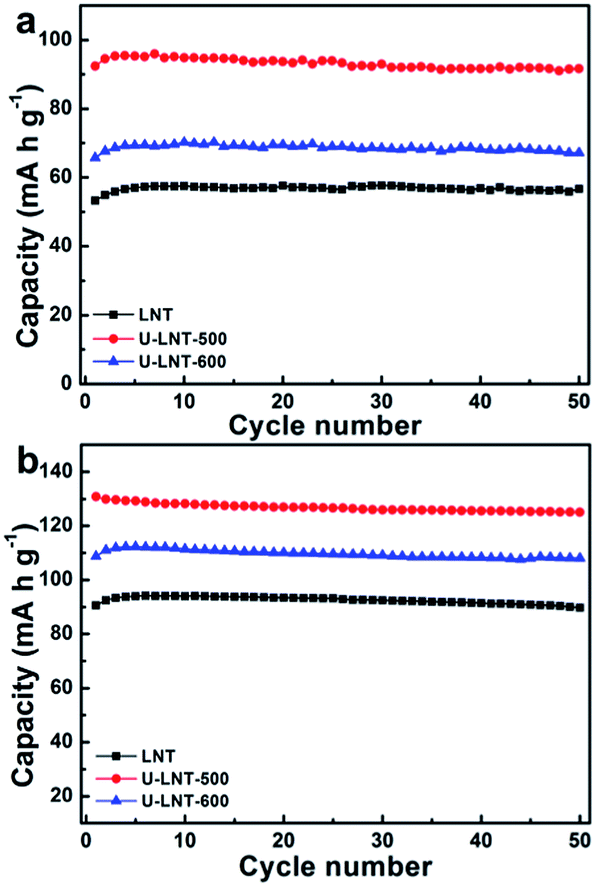 | ||
| Fig. 4 The cyclic performances (a) at ambient temperature and (b) at 50 °C. | ||
All of the three samples also appear outstanding cycling behaviours at 50 °C in Fig. 4b. The capacities for all samples at high temperature are higher than those measured at ambient temperature at the same rate. LNT delivered an initial discharge capacity of 54 mA h g−1 at ambient temperature, while 91 mA h g−1 is delivered at 50 °C, increasing about 69%. For U-LNT-500, the initial discharge capacity operated at 50 °C is 130 mA h g−1, 41% higher than the value operated at the ambient temperature. In the case of the U-LNT-600 sample, the initial capacity increment is 63% when the temperature increases to 50 °C. These results suggest that the untreated sample is prone to be affected by the operating temperature compared to the treated ones, implying that its poor electrochemical properties are controlled by the kinetic factors. It is worth mentioning that the discharge capacity of U-LNT-500 at ambient temperature is almost as high as the discharge capacity of LNT at 50 °C, suggesting that the urea treatment significantly enhance electrode reaction activity.
Urea treatment has beneficial effect not only on the discharge capacity, but also on coulombic efficiency (CE) at both ambient temperature and 50 °C (in Fig. 5). At the two temperatures, the trend of improvement for CE is similar and the differences of CE among the three samples are mainly in the first two cycles. At ambient temperature, the CEs of LNT in the first two cycles are 65.2% and 92.3%, respectively, while CEs are 71.8% and 98.4% for U-LNT-500, increasing about 6% separately. Therefore, the CE was clearly improved for U-LNT-500. For U-LNT-600, the improvement is much smaller, When compared with U-LNT-500. Therefore, the better urea treatment temperature should be 500 °C, and the reversibility of lithium insertion/deinsertion was increased after urea treatment.
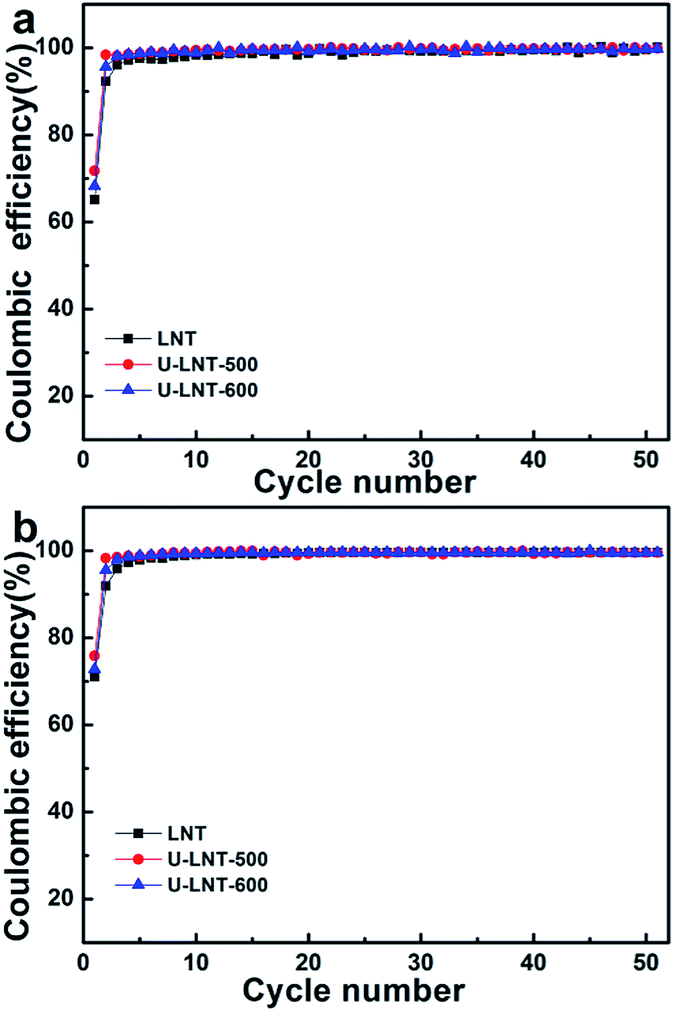 | ||
| Fig. 5 Coulombic efficiencies (percent discharge–charge capacity ratio) obtained at 0.5 C (a) at ambient temperature and (b) at 50 °C. | ||
To study how the urea affects the sample electrochemical performance, the N2 adsorption–desorption isotherms and the pore size distributions were performed. The specific surface areas and mean pore sizes for the three samples were listed in Table 2. The nitrogen sorption isotherms (Fig. 6a) of the three samples can be identified as the typical type V isotherms with a H3-type hysteresis loop. The hysteresis loop in the relative (P/P0) range of 0.4–1 may be ascribed to the presences of slit pores as well as mesopores, which can also be confirmed by Barrett–Joyner–Halanda (BJH) pore-size distribution (Fig. 6b). In Fig. 6b, the narrow distribution, mainly in the range from 0 to 13 nm, reveals the existence of slit pores. The slit pores among 2–13 nm and the wide distribution around 24 nm indicate the presence of mesopores. In Fig. 6b, the major mesopore size of U-LNT-500 falls in the range of 20–34 nm, which cause that the mean pore size of U-LNT-500 is expanded to 23.7 nm from 4.2 nm of LNT and the specific surface area is also enlarged to 15 m2 g−1 from 10 m2 g−1, correspondingly. The higher specific surface area and bigger pore size can facilitate the interface electrochemical reaction between electrolyte and electrode, and also shorten diffusion paths for electrons and ions. As a result, electrochemical performance was improved.
| Sample | SA (m2 g−1) | D (nm) |
|---|---|---|
| LNT | 10 | 4.2 |
| U-LNT-500 | 15 | 23.7 |
| U-LNT-600 | 12 | 3.4 |
 | ||
| Fig. 6 (a) N2 adsorption–desorption isotherm curves and (b) BJH pore size distribution plots of LNT, U-LNT-500, and U-LNT-600. | ||
TEM and HRTEM were displayed in Fig. 7. As the TEM image revealed in Fig. 7a, LNT sample has a particle-agglomerated morphology and the primary particles size is about 50 nm. Moreover, every particle is solid, thus the formation of the pore is piled by these primary particles, which are very well accordance with the nitrogen sorption isotherms analysis. As depicted in Fig. 7b, some faint lattice fringes and some bright areas, denoting the mixing of metal ions in the LNT particles, can be observed from the close observation. These are corresponded to with the XRD analysis, demonstrating that the Li2NiTiO4 pertained to disordered rock salt structure. For U-LNT-500 (Fig. 7c), close observation of the lattice arrays reveals that there are many domains where the lattice fringes are ordered arranged and the bright areas disappear. Combined with XPS that suggested the urea only had effect on the Ni valence, we deduce that the Li and Ni ions may rearranged in the urea treating circumstance so that LNT becomes an local ordered structure after urea treatment at 500 °C. This ordered structure provides facile diffusion tunnels inside the material for Li-ions and electrons transport and enhances lithium ions diffusion kinetics characteristic of Li2NiTiO4. In the case of the U-LNT-600 sample (Fig. 7d), its lattice fringes become crisscrossed arrangement, different from the patterns of both LNT and U-LNT-500. We believe that the higher urea treatment temperature may give rise to the loss of Li-ions leading to the presence of Ni3+ detected by XPS, thus the structure ordering property is disturbed. Therefore, combined with the smallest mean pore size of 3.4 nm in Table 2, U-LNT-600 exhibited a slightly degradation in terms of its electrochemical performances, when compared with U-LNT-500.
 | ||
| Fig. 7 TEM micrograph of (a) LNT; HRTEM images of (b) LNT, (c) U-LNT-500, and (d) U-LNT-600. | ||
4. Conclusions
Disordered Li2NiTiO4 nano-particles were synthesized by sol–gel method. At the low synthesis temperature, Ni ions had two coexistence forms: Ni2+ and Ni3+, which brought about the smaller lattice constant and volume. To improve the capacity and rate performance, urea was used to treat this material. Compared with LNT, the capacity, structure reversibility and rate capability for the treated sample are all remarkably improved and the inner electrode resistance is reduced effectively as well. We believed that the improved electrochemical performances after urea treatment should be resulted from the increase of the specific surface area and the bigger pore size as well as the conversion of the local ordered structure.Acknowledgements
This work was financially supported by the Ministry of Science and Technology of People’s Republic of China (contract number: 2010EG111015) and Science & Technology Office of Jiangsu Province (BE2013006-3).Notes and references
- M. Rosa Palacín, Chem. Soc. Rev., 2009, 38, 2565–2575 RSC.
- N.-S. Choi, Z. H. Chen, S. A. Freunberger, X. L. Ji, Y. K. Sun, K. Amine, G. Yushin, L. F. Nazar, J. Cho and P. G. Bruce, Angew. Chem., Int. Ed., 2012, 51, 9994–10024 CrossRef CAS PubMed.
- A. Manthiram, J. Phys. Chem. Lett., 2011, 2, 176–184 CrossRef CAS.
- S. Jeong, S. Park and J. Cho, Adv. Energy Mater., 2011, 1, 821–828 CrossRef.
- H. X. Deng, I. Belharouak, Y. K. Sun and K. Amine, J. Mater. Chem., 2009, 19, 4510–4516 RSC.
- D. Liu, W. Zhu, J. Trottier, C. Gagnon, F. Barray, A. Guerfi, A. Mauger, H. Groult, C. M. Julien, J. B. Goodenough and K. Zaghib, RSC Adv., 2014, 4, 154–167 RSC.
- A. R. Armstrong, M. Holzapfel, P. Novák, C. S. Johnson, S.-H. Kang, M. M. Thackeray and P. G. Bruce, J. Am. Chem. Soc., 2006, 128, 8694–8698 CrossRef CAS PubMed.
- L. Baggetto, D. Mohanty, R. A. Meisner, C. A. Bridges, C. Daniel, D. L. Wood III, N. J. Dudney and G. M. Veith, RSC Adv., 2014, 4, 23364–23371 RSC.
- L. Sebastian and J. Gopalakrishnan, J. Solid State Chem., 2003, 172, 171–177 CrossRef CAS.
- R. Trócoli, M. Cruz-Yusta, J. Morales and J. Santos-Pena, Electrochim. Acta, 2013, 100, 93–100 CrossRef PubMed.
- M. Kuezma, R. Dominko, D. Hanžel, A. Kodre, I. Arčon, A. Meden and M. Gaberšček, J. Electrochem. Soc., 2009, 156, A809–A816 CrossRef CAS PubMed.
- S. R. S. Prabaharan, M. S. Michael, H. Ikuta, Y. Uchimoto and M. Wakihara, Solid State Ionics, 2004, 172, 39–45 CrossRef CAS PubMed.
- M. Kuezma, R. Dominko, A. Meden, D. Makovec, M. Bele, J. Jamnik and M. Gaber-scek, J. Power Sources, 2009, 189, 81–88 CrossRef CAS PubMed.
- Y. Kawano, A. Kitajou and S. Okada, J. Power Sources, 2013, 242, 768–774 CrossRef CAS PubMed.
- H. Ming, J. Ming, X. W. Li, Q. Zhou, H. H. Wang, L. L. Jin, Y. Fu, J. Adkins and J. W. Zheng, Electrochim. Acta, 2014, 116, 224–229 CrossRef CAS PubMed.
- Y. Wang, H. Zhang, W. H. Chen, Z. Y. Ma and Z. C. Li, RSC Adv., 2014, 4, 37148–37156 RSC.
- Y. Zhao, Y. E. L. Fan, Y. Qiu and S. Yang, Electrochim. Acta, 2007, 52, 58–73 Search PubMed.
- G. Singh, R. Thomas, A. Kumar, R. S. Katiyar and A. Manivannan, J. Electrochem. Soc., 2012, 159, A470–A478 CrossRef CAS PubMed.
- X. Zhao, Y. J. Cui, L. Xiao, H. X. Liang and H. X. Li, Solid State Ionics, 2011, 192, 321–325 CrossRef CAS PubMed.
- S. J. Richard Prabakar, S. C. Han, S. P. Singh, D. K. Lee, K. S. Sohn and M. Pyo, J. Power Sources, 2012, 209, 57–64 CrossRef PubMed.
- Z. N. Wan, R. Cai, S. M. Jiang and Z. P. Shao, J. Mater. Chem., 2012, 22, 17773–17781 RSC.
- S. Z. Hu, A. J. Wang, X. Li and H. Lowe, J. Phys. Chem. Solids, 2010, 71, 156–162 CrossRef CAS PubMed.
- X. B. Chen, L. Liu, P. Y. Yu and S. S. Mao, Science, 2011, 331, 746–750 CrossRef CAS PubMed.
- G. M. Wang, H. Wang, Y. Ling, Y. Tang, X. Yang, R. C. Fitzmorris, C. Wang, J. Z. Zhang and Y. Li, Nano Lett., 2011, 11, 3026–3033 CrossRef CAS PubMed.
- X. W. Lou, D. Deng, J. Y. Lee and L. A. Archer, Chem. Mater., 2008, 20, 6562–6566 CrossRef CAS.
- L. Q. Zhang, H. Noguchi, D. C. Li, T. Muta, X. Q. Wang, M. Yoshioa and I. Taniguchi, J. Power Sources, 2008, 185, 534–541 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |