
Synergistic effect of C, Ag-codoped TiO2 photocatalyst within the GGA + U framework
Meili Guo*
Department of Physics, School of Science, Tianjin Chengjian University, Tianjin, 300384, China. E-mail: meiliguo314@163.com
First published on 12th November 2014
Abstract
The electronic structures and optical properties of C, Ag-codoped TiO2 were investigated within the GGA + U framework. Different doping concentration and different dopant distance (CsAgs (near and far), C2sAgs (near and far), and C3sAgs (near and far)) models were constructed. According to the formation energy results of these codoped systems, it was found that low concentration codoping was more stable than high concentration codoping. Therefore, we chose the CsAgs (near and far) codoped TiO2 to calculate further electronic structures and optical properties. It was found that near distance codoping could narrow the band gap to 2.81 eV, and far distance codoping decreased the band gap to 2.66 eV. Meanwhile, it was found that far distance codoping preferred to form more localized states near the top of the valence band, while near distance codoping could induce more significant hybrid states in the band gap. Optical property showed that C and Ag codoping could induce a synergistic effect compared with single C and single Ag doping. After codoping, the visible absorption was stronger for both the near and far distance codoped systems than those of the single doped systems, moreover, the far distance codoping configuration could induce both more significant band edge shift and stronger visible optical absorption than that of the near distance codoping configuration.
Introduction
TiO2 is a highly efficient photocatalytic material for energy and environmental applications due to its strong oxidizing power, high chemical inertness, low cost, and long-term stability.1–3 Unfortunately, the band gap of TiO2 is large (3.2 eV for anatase and 3.0 eV for the rutile structure), and absorbs very little visible light, which prevents its wide application in the visible region.4 To utilize solar light, it is necessary to enhance the visible optical absorption of TiO2.Band gap narrowing and introducing gap states are two effective routes to improve the visible optical absorption of TiO2. Nonmetal doping can create electronic states near the valence band or behave as part of the valence band, and thus can narrow the band gap of TiO2.5–8 For example, N doping is an effective route to create N 2p electronic states near the top of the valence band, and lots of experiments have confirmed the enhancement of the visible light response of N-doped TiO2.9–15 In addition, C-doped TiO2 showed 0.12–0.3 eV 2p electronic states near the top of the valence band, and X-ray photoelectron spectroscopy showed an increased electron density of states above the valence band of TiO2.16–18 In contrast, metal doping prefers to produce electronic states in the band gap, and thus induce the visible optical absorption centre.19–21 It has also been shown that metal-doped TiO2 has visible photocatalytic properties.22,23
Due to the individual benefits of metal and nonmetal doping, it has been proposed that metal and nonmetal codoping can combine the two benefits and thus can induce some interesting results.24 It was reported that codoping could improve the photoactivity of TiO2 by introducing gap states and band gap narrowing.24–29 For example, Gai et al. calculated the band structure and electronic state density of metal and nonmetal codoped TiO2 in detail, and they predicted C, Mo-codoped TiO2 could be conceived as a visible photocatalyst.28 The band edges of TiO2 could be modified by passivated C, Mo-codopants to shift the valence band edge up significantly, while leaving the conduction band edge almost unchanged, thus satisfying the stringent requirements. Subsequently, the experiment confirmed that C, Mo-codoping could enhance the photocatalytic activity of TiO2.30 In another independent work, Zhu et al. reported that noncompensated n–p codoping effectively narrows the band gap, and N, Cr-codoping could be a good candidate for enhancing the visible photoactivity of TiO2.31,32
In addition, atom configuration can play a very important role in a codoped system. For a codoped system, the far distance configuration of the doping atoms can avoid the formation of hybrid electronic states,33 and will benefit band gap engineering. However, sometimes, some hybrid electronic states caused by near distance doping can form some special structures which could induce some unexpected optical phenomena and thus improve the visible photoactivity.34 For example, for the N, B-codoped system, the electronic states at the top of the valence band and in the band gap will be significantly different for different doping distances.35,36 Besides, it was reported that the distance between dopant atoms in carbon nanotubes had a significant effect on the electronic structure.37 Thus, we conceived that different doping configurations will have a significant influence on the electronic structure and optical properties of codoped TiO2, which can provide some information for the development of visible photocatalytic and water splitting materials. In this work, in order to guarantee the reliability of the results, we constructed models with different doping concentrations and different dopant distance (CsAgs (near and far), C2sAgs (near and far), and C3sAgs (near and far)), however, the formation energy results indicated that the configuration with a high codoping concentration was unstable. Therefore, we presented the electronic structures and optical properties of C, Ag-codoped anatase TiO2 with the low doping concentration (CsAgs (near and far)). Meanwhile, the GGA + U method was employed to correct the band gap.
Computational methods
All the calculations were based on density functional theory (DFT), within the plane-wave-pseudo-potential approach, together with the Perdew–Burke–Ernzerhof (PBE) exchange correlation functional.38,39 The interaction between the valence electrons and the ionic core was described by ultrasoft pseudopotential, which used 2s22p4, 3d24s2, 2s22p2 and 4d105s1 as the valence electron configurations for the O, Ti, C, and Ag atoms, respectively. We chose the energy cut off of 340 eV for the pure and C, Ag-codoped TiO2 systems. The Brillouin-zone sampling mesh parameters for the k-point set were 1 × 2 × 2.40Firstly, geometry optimization was performed by the GGA method, because the geometry parameters calculated by the GGA method are closer to the experiments than those calculated by the GGA + U method.41 In this way, atomic positions and lattice parameters were optimized. The equilibrium lattice constants and fractional atomic coordinates were deduced from the total-energy minimization using the BFGS geometry optimization method. Relaxation of the lattice parameters and atomic positions was carried out under the constraint of the unit-cell space group symmetry. Energy–volume relationships were obtained by varying the unit-cell volume and the fitted results were obtained using the Murnaghan equation of state:
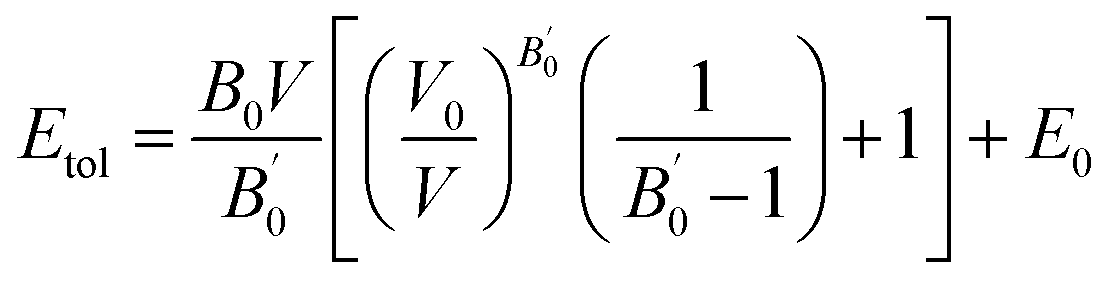
From the fitted results, the estimate of the static bulk modulus B0 at zero pressure, and the first-order pressure derivative of the bulk modulus B′0 were obtained. In the optimization process, the energy change, maximum force, maximum stress and maximum displacement tolerances were set as 2 × 10−5 eV per atom, 0.05 eV Å−1, 0.1 GPa and 0.002 Å, respectively.
Then, the electronic structure and optical properties of C, Ag-codoped TiO2 were calculated using the GGA + U method.42 The GGA + U approach introduces an intra-atomic electron–electron interaction as an on-site correction in order to describe systems with localized d states, which can produce a better band gap relative to the GGA method.43 In previous work, we calculated the U value dependent band gap, and found that the band gap with U = 6.6 eV was close to the real band gap and also didn’t induce unphysical results.44 Meanwhile, this U value has also been tested by another group.45 Thus, to account for the strongly correlated interactions of the Ti 3d electrons, a moderate on-site Coulomb repulsion U = 6.6 eV was applied to the further calculations of the electronic structures and optical properties in the present work.46 All parameters, such as k-point and the cut off energy were the same as the structural optimization.
To calculate the electronic structures and optical properties of pure and C, Ag-codoped anatase TiO2, a supercell was used, which contained 72 atoms. The substitution method was taken into account in this paper. Our study was based on a supercell with 72 atoms, as a 2 × 3 × 1 repetition of the primitive anatase unit cell. For the C, Ag-codoped anatase TiO2, different numbers of O atoms were replaced with C atoms, and one Ti atom was replaced with a Ag atom. Substitution models were formed with configurations of Ti23O47C1Ag1, Ti23O46C2Ag1, and Ti23O45C3Ag1. Substitution models are presented in Fig. 1.
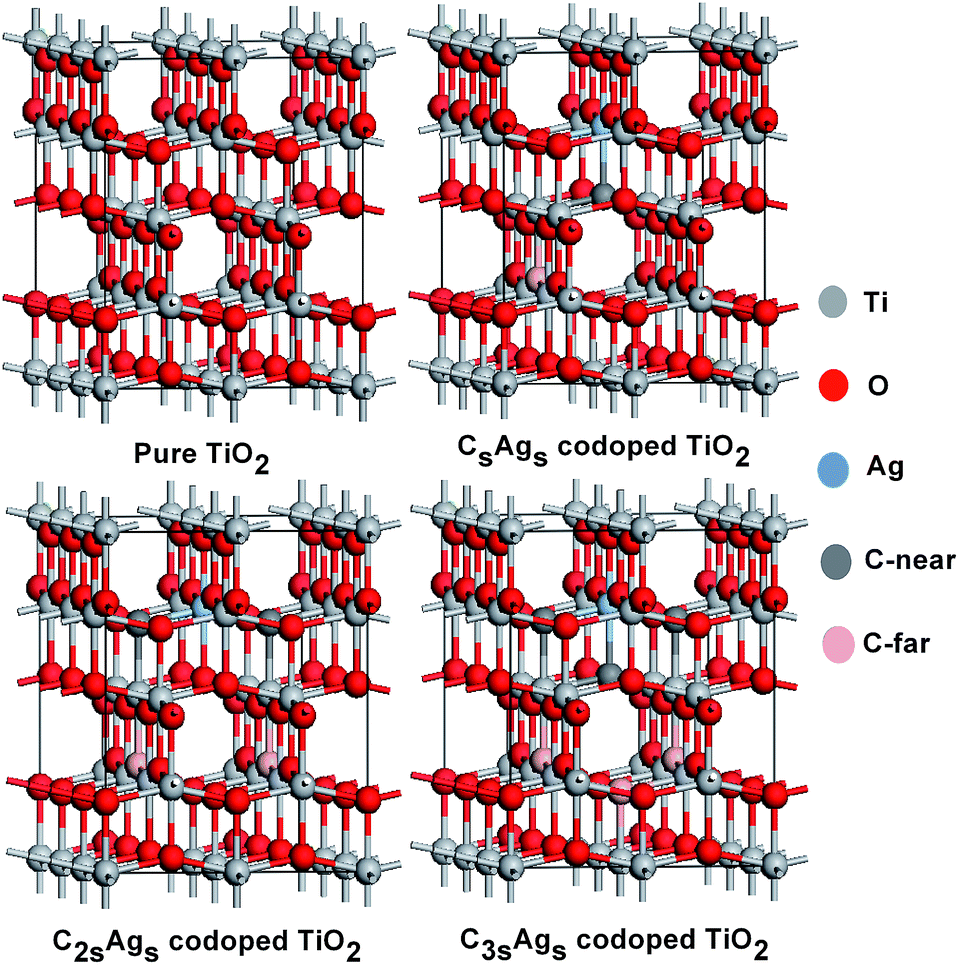 | ||
| Fig. 1 Pure and substitution models of CsAgs, C2sAgs, and C3sAgs-codoped TiO2. | ||
Results and discussion
To evaluate the feasibility of codoped systems, studies on the optimization, lattice distortion, and formation energies were carried out. We first checked the structural properties of CsAgs, C2sAgs, and C3sAgs-codoped TiO2. Table 1 gives the lattice parameters a and c of the pure and codoped TiO2 systems. For pure TiO2, the lattice constants were 3.776 Å for a and 9.486 Å for c, which were very close to the other calculation result and the experimental data.47–49 After doping, a and c were 3.815 and 9.845 Å for the far CsAgs-codoped TiO2, and 3.673 and 10.390 Å for the near CsAgs-codoped TiO2, respectively. Furthermore, the lattice constant a increased to 3.941 and 3.959 Å for the far C2sAgs and C3Ags-codoped TiO2, but decreased to 3.648 and 3.638 Å for the near C2sAgs and C3Ags-codoped TiO2. Generally, it could be seen that the far distance codoping induced less lattice distortion than that of the near distance codoping. Meanwhile, the CsAgs-codoping suffered less lattice distortion at a low concentration of C. However, with the increase of C doping concentration, c increased sharply. This indicates that high concentration may induce large lattice distortion and potential instability.| a (Å) | c (Å) | |
|---|---|---|
| Pure TiO2 | 3.776 | 9.486 |
| CsAgs far | 3.815 | 9.845 |
| CsAgs near | 3.673 | 10.390 |
| C2sAgs far | 3.941 | 10.042 |
| C2sAgs near | 3.648 | 10.591 |
| C3sAgs far | 3.959 | 10.146 |
| C3sAgs near | 3.638 | 10.691 |
Then, we calculated the formation energy. For a supercell, the formation energy of a doped system is defined as:41,50–52
| Ef = E(codoped) − E(pure) − nμC − μAg + nμO + μTi |
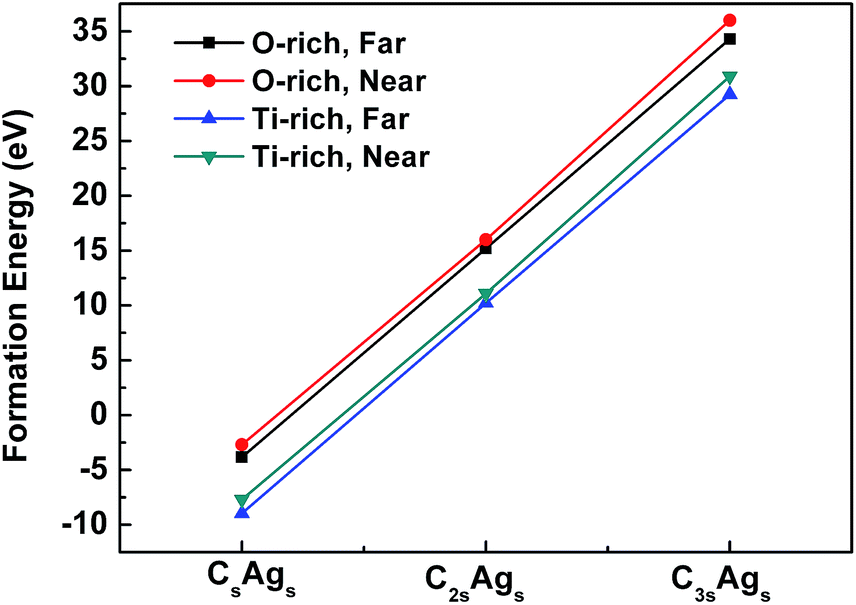 | ||
| Fig. 2 Formation energies of CsAgs, C2sAgs, and C3sAgs-codoped TiO2 under Ti-rich and O-rich conditions. | ||
We then moved to the energy band structure calculation. Fig. 3 shows the band structures of the near and far CsAgs-codoped TiO2. It was shown that the band gap of the near CsAgs-codoped TiO2 was 2.81 eV, which was lower than 2.89 eV of pure TiO2. Meanwhile, doped states could be found in the band gap. In contrast, the band gap of the far CsAgs-codoped TiO2 was decreased to 2.66 eV. Similarly, some discrete energy could also be found in the band gap. These band gap states are close to previously reported codoped systems, such as N, Cr-codoped TiO2.53 It is necessary to investigate the electronic states in the gap in detail.
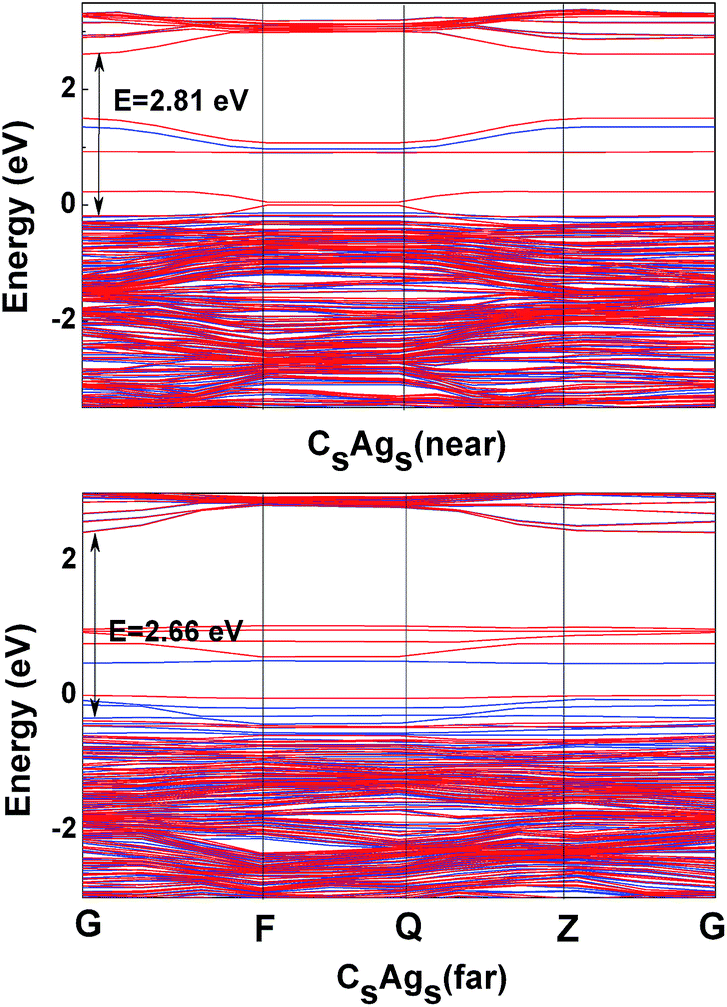 | ||
| Fig. 3 Energy band structure of the near and far CsAgs-codoped TiO2. The blue line represents up spin and the red line represents down spin. | ||
To explore the electronic states of the pure, near and far distance CsAgs-codoped TiO2, we calculated the density of states (DOS), which are presented in Fig. 4. Compared with pure TiO2 (Fig. 4a), C and Ag codoping could induce mid-gap states in both the spin-up and spin-down bands at low C concentration, which were mainly composed of the C 2p and Ag 4d electronic states in the valence band maximum (Fig. 4b). The energy level of the middle states was deeper for the near distance codoping. Meanwhile, both the far and near distance codoping induced some C 2p states to behave as part of the valence band, which was consistent with previous work. It has been widely reported that C 2p states could enhance the electronic states near the valence band.45 In addition, spin asymmetry could be observed for the codoped system, but was more significant for the far codoped system.
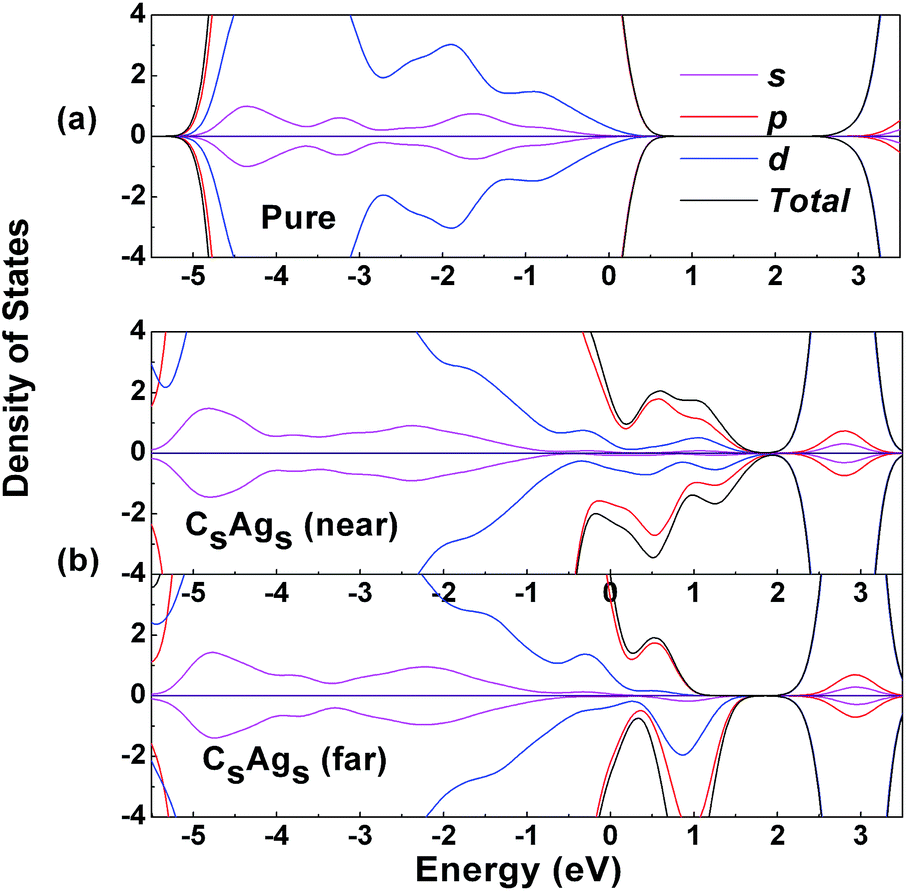 | ||
| Fig. 4 DOS of pure and CsAgs-codoped TiO2. | ||
To investigate the partial density of states (PDOS) of the CsAgs-codoped system, we further analyzed the Ag 4d and C 2p states (Fig. 5). It was clear that the C 2p of the near CsAgs-codoped TiO2 showed very good spin symmetry. In contrast, the C 2p from the far codoped TiO2 showed significant spin asymmetry. Meanwhile, Ag 4d states from the near codoped TiO2 also showed better spin symmetry than that of the far codoped TiO2. It could be concluded that the far distance codoping induced the significant spin asymmetry of C 2p and Ag 4d.
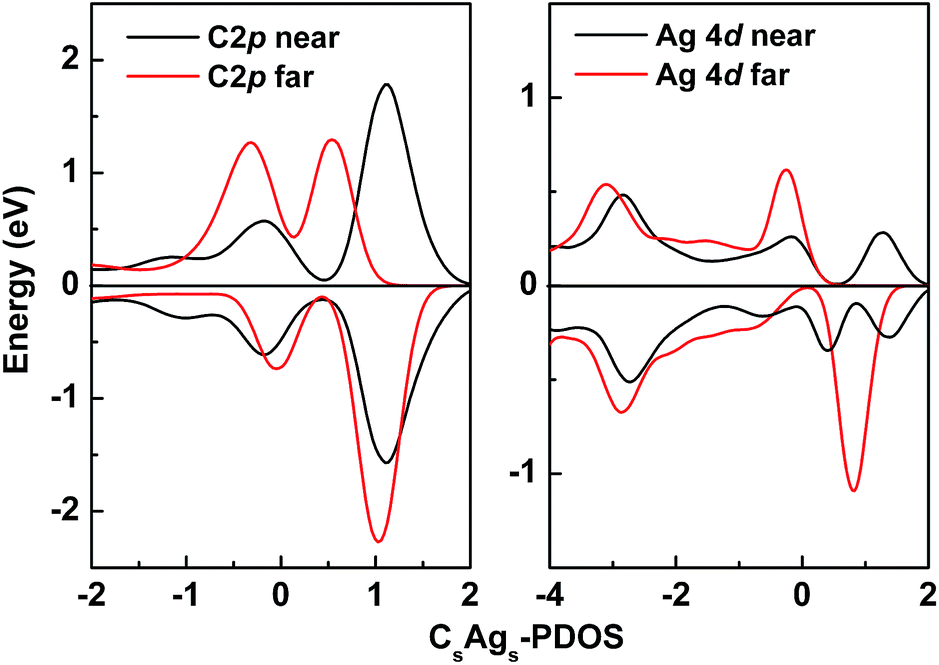 | ||
| Fig. 5 PDOS of the far and near CsAgs-codoped TiO2. | ||
Band gap narrowing was observed for the near and far distance CsAgs-codoped systems. The nature of this phenomenon was similar. Combining band structure and DOS, mid-gap states and band gap narrowing were both observed in the two codoped systems. However, for the near distance codoped system, the middle states were wider and deeper in gap. For the far distance codoped system, in contrast, the middle states were more narrow and induced a smaller band gap. Inspired by the electronic structure, it was of interest to further investigate the optical properties.
To investigate the optical transition of CsAgs-codoped TiO2, it is necessary to investigate the imaginary part of the dielectric function (ε2), because ε2 is important for describing the optical properties of any material. Optical transitions between occupied and unoccupied states are caused by the electric field of the photon. The spectra from the excited states can be described as a joint DOS between the valence and conduction bands. Optical transition peaks correspond to optical transitions between two states, and the intensity of the peaks is proportional to the density of states. For the pure TiO2, the 4.29 eV optical transition (Eg) was observed only (Fig. 6). After C and Ag codoping, the optical transition from the band gap decreased to 3.92 eV for the far distance C, Ag-codoped system, and 4.07 eV for the near distance C, Ag-codoped system. The redshift of the optical transition indicated that the band gaps of the CsAgs-codoped systems were decreased, which was in good agreement with the result of DOS. Moreover, the optical transition peak around 2 eV could be observed for the CsAgs-codoped TiO2 systems, which indicated that C and Ag codoping could induce a visible optical transition. The optical transition peak at 0.4 eV was observed for the near distance C, Ag-codoped TiO2, but this peak does not contribute to the visible light response. In addition, the optical properties of the single C and single Ag doped TiO2 were also investigated. It was found that C and Ag codoping could produce a synergistic effect which could further increase the visible absorption compared with single doping. Especially, the far distance codoping showed a stronger intensity visible optical transition compared to the near distance codoping, which was interesting. It is necessary to investigate the visible absorptions of the codoped systems.
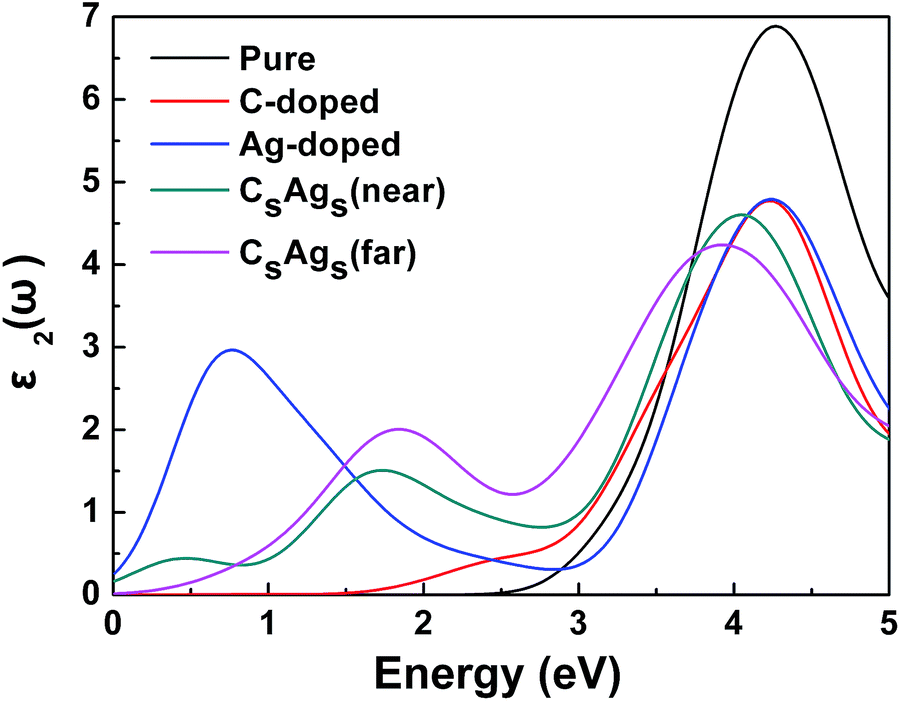 | ||
| Fig. 6 Imaginary parts of the dielectric function of pure, single C doped, single Ag doped, and CsAgs-codoped TiO2. | ||
Fig. 7 presents the optical absorptions of pure, single C doped, single Ag doped, and CsAgs-codoped TiO2. It was shown that the optical band edges of the single C doped and CsAgs-codoped TiO2 shifted to the visible light region compared to pure TiO2. It is well known that the relationship between the optical band gap and the absorption coefficient is given by54,55
| αhv = c(hv − Eg)1/2 |
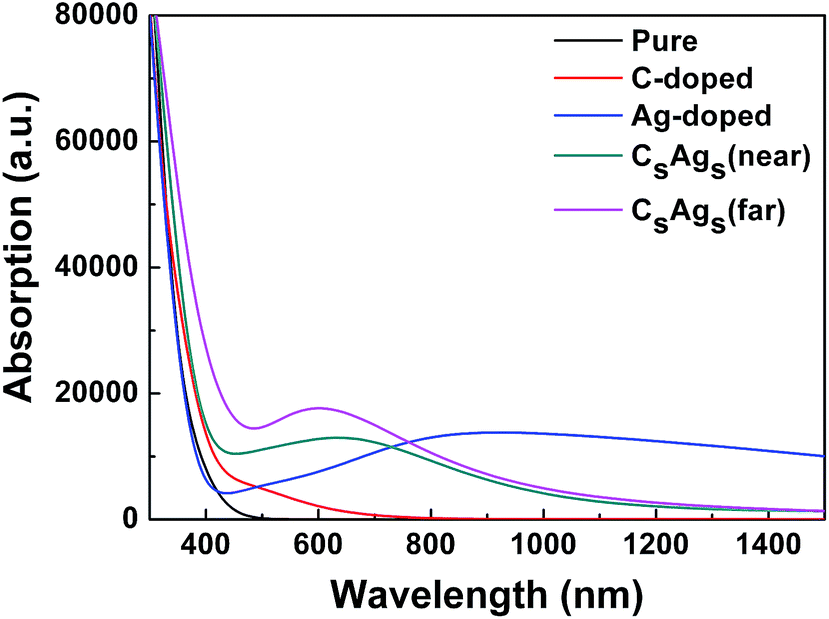 | ||
| Fig. 7 Optical absorption of pure, single C doped, single Ag doped, and CsAgs-codoped TiO2. | ||
Conclusion
In summary, the electronic structures and optical properties of single C doped, single Ag doped, and C, Ag-codoped TiO2 were calculated based on the GGA + U method. It was found that both far and near distance codoping induced band gap narrowing and visible optical absorption, and C and Ag codoping could produce an obvious synergistic effect compared with single C and single Ag doping. After codoping, visible absorption in the 450–800 nm range was sharply increased, moreover, the far distance codoping induced more significant band gap narrowing and visible optical absorption. Therefore, it is conceivable that C, Ag-codoped TiO2 would be useful for visible photoactivity.Acknowledgements
The author would like to thank Prof. Yanni Li, School of Chemical Engineering and Technology at Tianjin University, for providing the computational plat. This work is supported by the National Natural Science Foundation of China (Grant no. 11304220).References
- A. L. Linsebigler, G. Lu and J. T. Yates Jr, Chem. Rev., 1995, 95, 735–758 CrossRef CAS.
- S. U. Khan, M. Al-Shahry and W. B. Ingler, Science, 2002, 297, 2243–2245 CrossRef CAS PubMed.
- S. Song, B. Y. Xia, J. Chen, J. Yang, X. Shen, S. Fan, M. Guo, Y. Sun and X. Zhang, RSC Adv., 2014, 4, 42598–42603 RSC.
- T. Umebayashi, T. Yamaki, H. Itoh and K. Asai, Appl. Phys. Lett., 2002, 81, 454–456 CrossRef CAS PubMed.
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269–271 CrossRef CAS PubMed.
- J. Yu, Q. Xiang and M. Zhou, Appl. Catal., B, 2009, 90, 595–602 CrossRef CAS PubMed.
- M. Guo, X. Zhang and C. Liang, Phys. B, 2011, 406, 3354–3358 CrossRef CAS PubMed.
- M. L. Guo, X. D. Zhang, C. T. Liang and G. Z. Jia, Chin. Phys. Lett., 2010, 27, 057103 CrossRef.
- K. Yang, Y. Dai and B. Huang, J. Phys. Chem. C, 2007, 111, 12086–12090 CAS.
- S. Hoang, S. P. Berglund, N. T. Hahn, A. J. Bard and C. B. Mullins, J. Am. Chem. Soc., 2012, 134, 3659–3662 CrossRef CAS PubMed.
- F. Spadavecchia, G. Cappelletti, S. Ardizzone, M. Ceotto and L. Falciola, J. Phys. Chem. C, 2011, 115, 6381–6391 CAS.
- R. Long and N. J. English, Appl. Phys. Lett., 2009, 94, 132102 CrossRef PubMed.
- M. Harb, P. Sautet and P. Raybaud, J. Phys. Chem. C, 2011, 115, 19394–19404 CAS.
- Z. Lin, A. Orlov, R. M. Lambert and M. C. Payne, J. Phys. Chem. B, 2005, 109, 20948–20952 CrossRef CAS PubMed.
- J. Tao, L. Guan, J. Pan, C. Huan, L. Wang, J. Kuo, Z. Zhang, J. Chai and S. Wang, Appl. Phys. Lett., 2009, 95, 06250 Search PubMed.
- J. Yu, P. Zhou and Q. Li, Phys. Chem. Chem. Phys., 2013, 15, 12040–12047 RSC.
- F. Dong, S. Guo, H. Wang, X. Li and Z. Wu, J. Phys. Chem. C, 2011, 115, 13285–13292 CAS.
- X. Chen and C. Burda, J. Am. Chem. Soc., 2008, 130, 5018–5019 CrossRef CAS PubMed.
- W. Choi, A. Termin and M. R. Hoffmann, J. Phys. Chem., 1994, 98, 13669–13679 CrossRef.
- X. Zhang, M. Guo, C. Liu, W. Li and X. Hong, Appl. Phys. Lett., 2008, 93, 012103 CrossRef PubMed.
- M. Guo and J. Du, Phys. B, 2012, 407, 1003–1007 CrossRef CAS PubMed.
- B. Liu, H. M. Chen, C. Liu, S. C. Andrews, C. Hahn and P. Yang, J. Am. Chem. Soc., 2013, 135, 9995–9998 CrossRef CAS PubMed.
- Y. Ni, Y. Zhu and X. Ma, Dalton Trans., 2011, 40, 3689–3694 RSC.
- S. In, A. Orlov, R. Berg, F. García, S. Pedrosa-Jimenez, M. S. Tikhov, D. S. Wright and R. M. Lambert, J. Am. Chem. Soc., 2007, 129, 13790–13791 CrossRef CAS PubMed.
- J. Zhang, Y. Wu, M. Xing, S. A. K. Leghari and S. Sajjad, Energy Environ. Sci., 2010, 3, 715–726 CAS.
- Y.-F. Li, D. Xu, J. I. Oh, W. Shen, X. Li and Y. Yu, ACS Catal., 2012, 2, 391–398 CrossRef CAS.
- H. Kato and A. Kudo, J. Phys. Chem. B, 2002, 106, 5029–5034 CrossRef CAS.
- Y. Gai, J. Li, S.-S. Li, J.-B. Xia and S.-H. Wei, Phys. Rev. Lett., 2009, 102, 036402 CrossRef.
- L. Mi, P. Xu, H. Shen, P.-N. Wang and W. Shen, Appl. Phys. Lett., 2007, 90, 171909 CrossRef PubMed.
- J. Zhang, C. Pan, P. Fang, J. Wei and R. Xiong, ACS Appl. Mater. Interfaces, 2010, 2, 1173–1176 CAS.
- W. Zhu, X. Qiu, V. Iancu, X.-Q. Chen, H. Pan, W. Wang, N. M. Dimitrijevic, T. Rajh, H. M. Meyer, M. P. Paranthaman, G. M. Stocks, H. H. Weitering, B. Gu, G. Eres and Z. Zhang, Phys. Rev. Lett., 2009, 103, 226401 CrossRef.
- M. E. Kurtoglu, T. Longenbach, K. Sohlberg and Y. Gogotsi, J. Phys. Chem. C, 2011, 115, 17392–17399 CAS.
- L. Huang, A. Rosa and R. Ahuja, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 075206 CrossRef.
- W.-J. Yin, S.-H. Wei, M. M. Al-Jassim and Y. Yan, Phys. Rev. Lett., 2011, 106, 066801 CrossRef.
- M. Guo, X.-D. Zhang and J. Du, Phys. Status Solidi RRL, 2012, 6, 172–174 CrossRef CAS.
- N. Feng, A. Zheng, Q. Wang, P. Ren, X. Gao, S.-B. Liu, Z. Shen, T. Chen and F. Deng, J. Phys. Chem. C, 2011, 115, 2709–2719 CAS.
- D. Carroll, P. Redlich, X. Blase, J.-C. Charlier, S. Curran, P. Ajayan, S. Roth and M. Rühle, Phys. Rev. Lett., 1998, 81, 2332 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS.
- M. C. Payne, M. P. Teter, D. C. Allan, T. Arias and J. Joannopoulos, Rev. Mod. Phys., 1992, 64, 1045–1097 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- R. Long and N. J. English, Chem. Phys. Lett., 2009, 478, 175–179 CrossRef CAS PubMed.
- L. Li, W. Wang, H. Liu, X. Liu, Q. Song and S. Ren, J. Phys. Chem. C, 2009, 113, 8460–8464 CAS.
- V. I. Anisimov, F. Aryasetiawan and A. Lichtenstein, J. Phys.: Condens. Matter, 1997, 9, 767–808 CrossRef CAS.
- M. Guo and J. Du, Int. J. Mod. Phys. B, 2013, 27, 1350123 CrossRef.
- K. Yang, Y. Dai, B. Huang and M.-H. Whangbo, J. Phys. Chem. C, 2009, 113, 2624–2629 CAS.
- H. J. Kulik, M. Cococcioni, D. A. Scherlis and N. Marzari, Phys. Rev. Lett., 2006, 97, 103001 CrossRef.
- S. Na-Phattalung, M. F. Smith, K. Kim, M.-H. Du, S.-H. Wei, S. Zhang and S. Limpijumnong, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 125205 CrossRef.
- A. Janotti, J. Varley, P. Rinke, N. Umezawa, G. Kresse and C. Van de Walle, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 085212 CrossRef.
- Y. Wang, H. Liu, Z. Li, X. Zhang, R. Zheng and S. Ringer, Appl. Phys. Lett., 2006, 89, 042511 CrossRef PubMed.
- F. Oba, A. Togo, I. Tanaka, J. Paier and G. Kresse, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 245202 CrossRef.
- J. Li, S.-H. Wei, S.-S. Li and J.-B. Xia, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 081201 CrossRef.
- X. Cui, J. Medvedeva, B. Delley, A. Freeman, N. Newman and C. Stampfl, Phys. Rev. Lett., 2005, 95, 256404 CrossRef CAS.
- M. Chiodi, C. P. Cheney, P. Vilmercati, E. Cavaliere, N. Mannella, H. H. Weitering and L. Gavioli, J. Phys. Chem. C, 2011, 116, 311–318 Search PubMed.
- X. Zhang, M. Guo, W. Li and C. Liu, J. Appl. Phys., 2008, 103, 063721 CrossRef PubMed.
- V. Srikant and D. R. Clarke, J. Appl. Phys., 1998, 83, 5447 CrossRef CAS PubMed.
- N. Feng, Q. Wang, A. Zheng, Z. Zhang, J. Fan, S.-B. Liu, J.-P. Amoureux and F. Deng, J. Am. Chem. Soc., 2013, 135, 1607–1616 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |