
A remarkable solvent effect on the reaction of 4-hydroxycoumarin with (E)-3-aryl-2-nitroprop-2-enol: Facile synthesis of highly substituted furo/pyrano[3,2-c]chromenes†
Shivendra Singh,
Anvita Srivastava,
Shaikh M. Mobin and
Sampak Samanta*
Indian Institute of Technology Indore, 452017, Indore, Madhya Pradesh, India. E-mail: sampaks@iiti.ac.in; sampak_s1@yahoo.com; Fax: +91-731-2364182; Tel: +91-731-2438742
First published on 10th December 2014
Abstract
A remarkable solvent effect on the reaction of 4-hydroxycoumarin derivatives with (E)-3-aryl/hetero-aryl-2-nitroprop-2-enols has been observed in water and DMSO media. This result was employed for the straightforward syntheses of new functionalized furo/pyrano[3,2-c]chromenes in 63–93% yields and diasteromeric ratio up to ≤99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Moreover, a simple, mild, efficient and catalyst-free one-pot method may offer an alternative synthetic strategy for annulating the furan/pyran rings on the coumarin nucleus. Furthermore, water has shown a significant positive effect on the rate and selectivity (product) of this reaction.
:1. Moreover, a simple, mild, efficient and catalyst-free one-pot method may offer an alternative synthetic strategy for annulating the furan/pyran rings on the coumarin nucleus. Furthermore, water has shown a significant positive effect on the rate and selectivity (product) of this reaction.
The development of a highly efficient protocol for the straightforward synthesis of functionalized furocoumarin scaffolds from simple raw materials is a key research area in synthetic organic chemistry and drug discovery programmes. A primary reason is that this principal core is frequently found in a broad range of natural products and active pharmacophores,1 many of them synthetic congeners showing potential biological activities2 (Fig. 1). Owing to the importance of these bioactive furocoumarin derivatives, several efforts have been devoted for the syntheses of substituted 4H-furo[3,2-c]chromen-4-one derivatives by adopting various modern techniques such as C–O and C–C bond coupling reactions,3a,c,d cascade reaction (Scheme 1a),3b,3e lactonization reaction3g using transition metal catalysts.3 However, all these protocols involve toxic/expensive metal catalysts, toxic reagents and harmful/hazardous organic solvents which are not much appreciable from environmental and economic stand points of view.
 | ||
| Fig. 1 Natural products and biologically active compounds that have a furocoumarin moiety. | ||
 | ||
| Scheme 1 Various approaches for furo[3,2-c]chromen-4-one derivatives. | ||
Besides, the great success of transition metal catalysts, a few examples of metal-free mediated one-pot synthesis of substituted furocoumarin derivatives have been well documented as some of these works were published very recently.4 For example, in 2012, Feist-Bénary/addition-elimination reaction of 4-hydroxycoumarin with nitroallyl acetate under basic conditions has been realized by Chen and co-workers (Scheme 1b).4b Afterwards, in 2013, Wang and co-workers also established a very attractive synthetic strategy involving a four-component reaction between β-nitrostyrene, aromatic aldehyde, 4-hydroxycoumarin and ammonium acetate under refluxing ethanol (Scheme 1c).4d Even with a noticeable progress, there is still no successful report on catalyst-free one-pot synthesis of 4H-furo[3,2-c]chromen-4-one derivatives in water medium. To address this synthetic challenge, we sought to devise an alternative practical, cost-effective and catalyst-free protocol for the preparation of new functionalized furocoumarin scaffolds from simple starting materials under aerobic conditions, specifically, in water which is still a preferred choice of synthetic organic and medicinal chemists.5
In recent years, performing the reaction with functionalized β-nitroolefins is an interesting subject for the access of various important heterocyclic scaffolds.6 In this regard, we also reported several methods for the syntheses of important class of functionalized heterocyclic compounds utilizing β-nitroolefins as Michael acceptors.7 Furthermore, we wish to report herein a mild, simple, convenient, high yielding procedure for the construction of 2-(hydroxymethyl)-3-aryl-4H-furo[3,2-c]chromen-4-one scaffolds via a one-pot reaction of substituted 4-hydroxycoumarins with (E)-3-aryl-2-nitroprop-2-enols in water under catalyst-free conditions (Scheme 1d).
The initial reaction between 4-hydroxycoumarin (1a) and E-3-phenyl-2-nitropro-2-enol (2a) was conducted in CHCl3 at 50 °C in the absence of catalyst. After 24 h, a trace amount of unexpected 2-(hydroxymethyl)-3-phenyl-4H-furo[3,2-c]chromen-4-one (3aa) along with trans-3-nitro-4-phenyl-3,4-dihydropyrano[3,2-c]chromen-5(2H)-one (4aa) were obtained in 12% and 3% yields respectively (entry 1, Table 1). Interestingly, when the same reaction was carried out at elevated temperature 70 °C, slight improvements of results were recorded in terms of reaction time (24 h to 15 h) and yield (3aa, 12–25%, entry 2). As we are aware that the reaction of 4-hydroxycoumarin with β-nitrostyrene derivatives highly depends on the polarity of the solvents8 which necessitates examining the reaction with different solvents in detail. Keeping in mind, we tested this reaction several common organic solvents namely EtOH, DMSO, DMF, CHCl3, 1,2-dichloroethane (DCE), toluene, MeCN at 70 °C. Pleasantly, we noticed that polar solvents like EtOH, DMF and DMSO favoured the formation of 4aa in good to high yields (42–85%, entries 6–8) and non-polar solvents (CHCl3, DCE, toluene and MeCN) led to the formation of furocoumarin 3aa in low yields (21–42%, entries 2–5). Screening of several catalysts (L-proline, DABCO, DMAP) revealed that they have almost no influence on product selectivity. However, they enhanced the rate of the reaction (5–10 h vs. 15 h), resulting in higher yields of 3aa (69–76%, entries 9–12) and 4aa (51–88% entries 13–18). In order to develop an environmental friendly reaction conditions, we performed this reaction in water instead of harmful organic solvent. Surprisingly, after 5 h, in the absence of catalyst, the reaction proceeded very smoothly at 70 °C, leading to the high yield of major product 3aa (83%, entry 19). In particular, there was no significant improvement of result in terms of yield, selectivity or time when the reaction was carried out in the presence of catalyst (L-proline, DABCO and DMAP) under identical conditions (entries 20–23). From the various reaction conditions as shown in Table 1, it was obvious that best result was obtained for 3aa at condition mentioned in entry 19 (83% yield).
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | Solvent | T (°C) | T h−1 | Yieldc (%) | |
| 3aa | 4aad | |||||
| a Unless otherwise specified, all the reactions were performed with compound 1a (0.25 mmol), 2a (0.3 mmol) and catalyst (0.05 mmol, 20 mol%) in specified solvent (0.6 mL) and temperature.b Nil indicates no catalyst.c Yield of isolated product after column chromatography.d Diastereomeric ratio (99:1) of the crude product was recorded by 1H NMR. |
||||||
| 1b | Nil | CHCl3 | 50 | 24 | 12 | <3 |
| 2b | Nil | CHCl3 | 70 | 15 | 25 | <5 |
| 3b | Nil | DCE | 70 | 15 | 21 | <7 |
| 4b | Nil | Toluene | 70 | 15 | 41 | 8 |
| 5b | Nil | MeCN | 70 | 15 | 29 | <7 |
| 6b | Nil | EtOH | 70 | 15 | 19 | 42 |
| 7b | Nil | DMSO | 70 | 15 | 6 | 85 |
| 8b | Nil | DMF | 70 | 15 | 11 | 81 |
| 9 | L-Proline | CHCl3 | 70 | 10 | 74 | 9 |
| 10 | L-Proline | DCE | 70 | 10 | 76 | <8 |
| 11 | L-Proline | Toluene | 70 | 10 | 72 | 11 |
| 12 | L-Proline | MeCN | 70 | 10 | 69 | 7 |
| 13 | L-Proline | DMSO | 70 | 5 | 7 | 88 |
| 14 | L-Proline | DMF | 70 | 7 | 10 | 83 |
| 15 | L-Proline | EtOH | 70 | 7 | 29 | 51 |
| 16 | DABCO | DMSO | 70 | 5 | 9 | 81 |
| 17 | DMAP | DMSO | 70 | 5 | 12 | 83 |
| 18 | DABCO | DMF | 70 | 7 | 13 | 77 |
| 19b | Nil | H2O | 70 | 5 | 83 | <6 |
| 20 | L-proline | H2O | 70 | 5 | 86 | 8 |
| 22 | DABCO | H2O | 70 | 5 | 83 | 9 |
| 23 | DMAP | H2O | 70 | 5 | 84 | <7 |

Herein we present the following probable mechanism for the formations of compounds 3aa and 4aa under present reaction conditions as shown in Scheme 2. In case of water medium, we assume that water plays a crucial role in this reaction by acting as an amphiphilic dual-catalyst.9 It may activate both the substrates 1a and 2a through intermolecular H-bonding9a which increases rate of the Michael addition reaction between 1a and 2a, resulting information of nitronic acid intermediate 5. Afterwards, the intermediate 6 is generated from 5 via a tautomerization process which undergoes in turn intramolecular cyclization, subsequent dehydration and elimination of hyponitrous acid (HNO) to give the final compound 3aa (Path A).
 | ||
| Scheme 2 Proposed mechanism for the formations of compounds 3aa and 4aa. | ||
On the other hand, the intermediate 5 instead tend to form an intermediate 8 under this condition via a tautomerization process, which is followed by intramolecular cyclization-dehydration, leading to the pyranocoumarin 4aa. In case of L-proline, we think at this point that Michael reaction may take place through an enamine intermediate 1b′ to form 1b′′ which upon reaction with water and followed by elimination of L-proline furnishes intermediate 5. The finally products 3aa and 4aa are generated from 5 by following path A and path B respectively. However, additional work is necessary for understanding the detailed mechanism of this reaction.
With the optimum reaction conditions in hand, various substituted (E)-3-aryl-substituted-2-nitroprop-2-enols and 4-hydroxycoumarin derivatives were examined to understand the generality and scope of this reaction. The outcomes are compiled in Table 2. It is noteworthy that both electron-donating (Me, MeO and OH) and electron-withdrawing (Cl, Br, and NO2) groups on the aromatic rings of 3-aryl-substitited-2-nitroprop-2-enols annulated smoothly with substrate (1a), providing the functionalized furo[3,2-c]chromen-4-ones (3aa–3aj) in good to high yields (63–88%, entries 2–10). Similarly, incorporation of several functional groups such as electron donating (Me) and electron withdrawing (Cl, Br and NO2) on aryl rings of 4-hydroxycoumarins (1b–e, entries 11–21) did not pose any with (E)-3-aryl-substituted-2-nitroprop-2-enols (2a–c and 2g, entries 11–21) by this procedure and resulted in clean and complete Michael/cyclization-elimination reactions, providing the corresponding annulated products (3ba–3ec) in good to high yields (63–88%). It is observed that 4-hydroxycoumarins (1c–e) possessing electron-withdrawing substituents are slightly less reactive than its un-substituted version 1a towards Michael acceptors under identical conditions (e.g. 5 h vs. 9 h, entry 1 vs. entry 20). Importantly, our present conditions are mild enough to retain various functional groups such as OMe, OH, Cl, Br, NO2, CH2OH etc.
|
|||||
|---|---|---|---|---|---|
| Entry | R1 | R | T h−1 | Name | Yieldb (%) |
| a All the reactions were done with 4-hydroxycoumarin derivative (1a–e, 0.25 mmol), (E)-3-aryl-2-nitropro-2-enols (2a–j, 0.3 mmol) in water (0.6 mL) at 70 °C.b Isolated yield of after column chromatography. | |||||
| 1 | H | Ph | 5 | 3aa | 83 |
| 2 | H | 4-MeC6H4 | 5 | 3ab | 84 |
| 3 | H | 4-MeOC6H4 | 5 | 3ac | 88 |
| 4 | H | 2-MeOC6H4 | 6 | 3ad | 87 |
| 5 | H | 3,4-(MeO)2C6H3 | 6 | 3ae | 82 |
| 6 | H | 2-HOC6H4 | 10 | 3af | 63 |
| 7 | H | 4-ClC6H4 | 5 | 3ag | 81 |
| 8 | H | 2-ClC6H4 | 5 | 3ah | 85 |
| 9 | H | 4-BrC6H4 | 5 | 3ai | 79 |
| 10 | H | 4-NO2C6H4 | 5 | 3aj | 83 |
| 11 | Me | Ph | 5 | 3ba | 86 |
| 12 | Me | 4-MeOC6H4 | 5 | 3bc | 88 |
| 13 | Me | 4-ClC6H4 | 5 | 3bg | 81 |
| 14 | Cl | Ph | 7 | 3ca | 79 |
| 15 | Cl | 4-MeC6H4 | 7 | 3cb | 80 |
| 16 | Cl | 4-ClC6H4 | 7 | 3cg | 77 |
| 17 | Br | Ph | 7 | 3da | 79 |
| 18 | Br | 4-MeC6H4 | 7 | 3db | 86 |
| 19 | Br | 4-ClC6H4 | 7 | 3dg | 78 |
| 20 | NO2 | Ph | 9 | 3ea | 63 |
| 21 | NO2 | 4-MeOC6H4 | 9 | 3ec | 67 |

Besides, the gram scale preparation of compound 3aa was investigated in our imposed conditions. A heterogeneous mixture of compound 1a (1.62 g, 10.0 mmol) and 2a (12.0 mmol) in water (6.0 mL) was heated at 70 °C for 6 h. Afterwards, water was decanted carefully from the reaction mixture to give the gummy residue which was directly purified by column chromatography technique, leading to the pure product 3aa in 76% yield. This interesting result reveals that our optimal condition can be used for milligram to gram scale synthesis.
Next, we turned our attention towards the facile synthesis of functionalized pyranocoumarin derivative as this motif is frequently existed in a variety of bioactive natural products and pharmacophores.10,11 Literature survey shows that several practical and efficient techniques are available for the syntheses of both the racemic and enantio-enriched versions of dihydropyrano[3,2-c]chromen derivatives.11 Towards our investigations, various functionalized 4-hydroxycoumarin derivatives (1a–b, 1e) were reacted with several aryl/hetero-aryl-substituted nitroallylic alcohols in DMSO medium at 70 °C in the presence of L-proline (20 mol%). The results are summarized in Scheme 3. To our delight, all the reactions proceeded smoothly by this procedure to furnish the corresponding previously unknown class of substituted trans-3-nitro-4-aryl-3,4-dihydropyrano[3,2-c]chromen-5(2H)-one derivatives in high to excellent yields (85–93%, 4aa–4ec) with excellent diastereoselectivities (up to ≤99:1 dr). It should be noted that relative stereochemistry of major diastereomer 4ah was unanimously confirmed by its single crystal X-ray diffraction data (Fig. 2, details in ESI†) indicating aryl group in trans-orientation with NO2. Similarly, several sensitive functional groups namely NO2, OMe, furan, Me and Cl are tolerable in our present conditions.
 | ||
| Scheme 3 One-pot synthesis of trans-3-nitro-4-aryl-3,4-dihydropyrano[3,2-c]chromen-5(2H)-ones. | ||
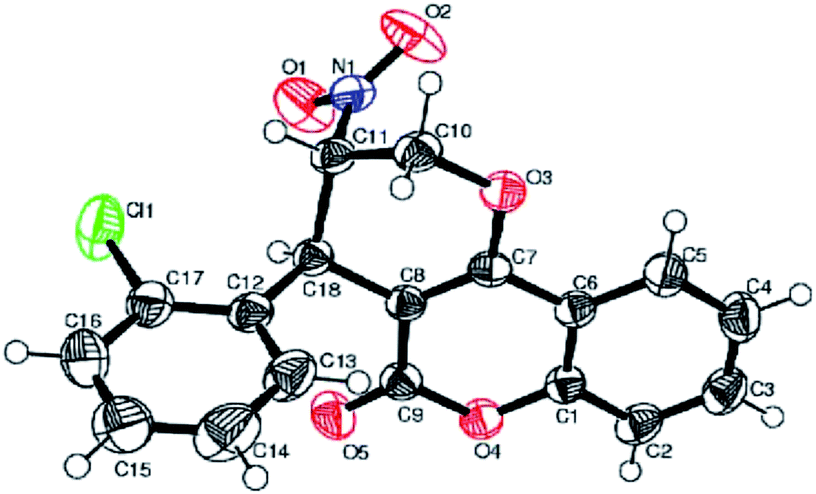 | ||
| Fig. 2 ORTEP diagram of major diastereomer 4ah, thermal ellipsoids drawn at the 50% probability level. | ||
In conclusion, we have observed a remarkable solvent effect on one-pot reaction of 4-hydroxycoumarin derivatives with (E)-3-aryl-substituted-2-nitroprop-2-enols and it was used for expeditious synthesis of highly substituted furo/pyrano[3,2-c]chromenes, by choosing water or DMSO as a solvent. Moreover, water has shown not only positive effects on the rate and selectivity (product) of this reaction but also beneficial features in terms of safety, health, cost-effectiveness and environmental points of view. Furthermore, our current procedure avoids the use of toxic metals and their salts, harmful/volatile organic solvents, expensive reagents, any need for dry conditions or inert-atmosphere, multi-step, etc. Importantly, in comparison to the established methods, our protocols are advantageous since they are operationally simple, easy to work-up, applicable for gram-scale synthesis, as well as offer good to excellent yields (63–93%), excellent diastereoselectivities (up to ≤99:1 dr) and good substrate scope. Further endeavours towards the detailed understanding of the reaction mechanism as well as applications of these compounds are under investigation and will be communicated in due course.
Acknowledgements
The authors thank CSIR (Project no. 02(0019)/11/EMR-II) and DST (Project No. SB/S1/OC-19/2013) research grants for the generous financial support respectively. S. S. and A. S. are also thankful to UGC for their fellowships.References
- Biologically active furocoumarin see:
(a) L. Santana, E. Uriarte, F. Roleira, N. Milhazes and F. Borges, Curr. Med. Chem., 2004, 11, 3239 CrossRef CAS
; (b) R. Gambari, I. Lampronti, N. Bianchi, C. Zuccato, G. Viola, D. Vedaldi and F. DallAÁcqua, Top. Heterocycl. Chem., 2007, 9, 265 CAS
-
(a) L. Piccagli, M. Borgatti, E. Nicolis, N. Bianchi, I. Mancini, I. Lampronti, D. Vevaldi, F. Dall'Acqua, G. Cabrini and R. Gambari, Bioorg. Med. Chem., 2010, 18, 8341 CrossRef CAS PubMed
-
(a) L. Chen, Y. Li and M.-H. Xu, Org. Biomol. Chem., 2010, 8, 3073 RSC
-
(a) C.-J. Lee, Y.-J. Jang, Z.-Z. Wu and W. Lin, Org. Lett., 2012, 14, 1906 CrossRef CAS PubMed
-
(a) M. B. Gawande, V. D. B. Bonifácio, R. Luque, P. S. Branco and R. S. Varma, Chem. Soc. Rev., 2013, 42, 5521 Search PubMed
-
(a) D. K. Nair, R. F. S. Menna-Barreto, E. N. da Silva Júnior, S. M. Mobin and I. N. N. Namboothiri, Chem. Commun., 2014, 50, 6973 RSC
-
(a) S. Biswas, P. K. Jaiswal, S. Singh, S. M. Mobin and S. Samanta, Org. Biomol. Chem., 2013, 11, 7084 RSC
-
(a) E. Ghabraie, M. Bararjanian, S. Balalaie, F. Rominger and H. R. Bijanzadeh, Helv. Chim. Acta, 2011, 94, 1440 CrossRef CAS
-
(a) D. K. Barange, V. Kavala, B. R. Raju, C.-W. Kuo, C. Tseng, Y.-C. Tu and C.-F. Yao, Tetrahedron Lett., 2009, 50, 5116 CrossRef CAS PubMed
- For biological and pharmaceutical activities of pyranocoumarin, see:
(a) R. D. H. Murray, J. Medez and S. A. Brown, The Natural Coumarins: Occurrence, Chemistry and Biochemistry, Wiley, New York, 1982 Search PubMed
-
(a) M. Rueping, E. Merino and E. Sugiono, Adv. Synth. Catal., 2008, 350, 2127 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization data of new compounds. CCDC 1011395. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ra10610e |
| This journal is © The Royal Society of Chemistry 2015 |