
Effect of the biobased linear long-chain monomer on crystallization and biodegradation behaviors of poly(butylene carbonate)-based copolycarbonates
Abstract
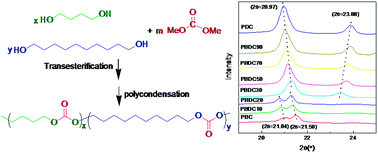
To improve the crystallization ability of poly(butylene carbonate) (PBC), a monomer with a linear long chain as a biobased derivative of castor oil was randomly introduced into the PBC main chain. A series of aliphatic copolycarbonates poly(butylene-co-decamethylene carbonate)s (PBDCs), with weight-average molecular weights of 125 000 to 202 000 g mol−1, were synthesized from dimethyl carbonate, 1,4-butanediol, and 1,10-decanediol via a two-step polycondensation process, using sodium acetylacetonate as the catalyst. The PBDCs, being statistically random copolymers, showed a single Tg over the entire composition range. The DSC results testified that the introduction of a decamethylene carbonate (DC) unit can significantly enhance the crystallization rate of PBC. The PBDC copolycarbonates had a minimum melting point in the plot of melting point versus composition. Wide-angle X-ray diffraction patterns showed that the copolycarbonates with up to 20 mol% DC units formed PBC type crystals, while those with higher DC unit content crystallized in poly(decamethylene carbonate) (PDC) type crystals. This indicates that the PBDC copolycarbonates show isodimorphic cocrystallization. The thermal stability, crystalline morphology, and enzymatic degradation of the PBDC copolycarbonates were also studied.
 Please wait while we load your content...
Please wait while we load your content...

