
Synthesis and click modification of an azido-functionalized Zr(IV) metal–organic framework and a catalytic study†
Xiu-Chun Yi,
Fu-Gui Xi,
Yan Qi and
En-Qing Gao*
Shanghai Key Laboratory of Green Chemistry and Chemical Processes, Department of Chemistry, East China Normal University, Shanghai 200062, P.R. China. E-mail: eqgao@chem.ecnu.edu.cn; Fax: +86-21-62233404; Tel: +86-21-62233404
First published on 21st November 2014
Abstract
An azido-functionalized Zr(II) metal–organic framework (MOF), UiO-67–N3, was synthesized from 2-azidobiphenyl-4,4′-dicarboxylic acid. During the synthesis, the ligand can undergo in situ thermocyclization to give 9H-carbazole-2,7-dicarboxylic acid. It proved that UiO-67–N3 can be obtained at relatively low temperature without ligand transformation. Post-synthetic modification of UiO-67–N3 was successfully performed via the click reactions between the azido group and different alkyne compounds to produce new MOFs with different functionalities, UiO-67–Tz–X with X = COOCH3, OH and NH2. These clicked MOFs, especially UiO-67–Tz–NH2, exhibit better stability than the mother material UiO-67–N3. The catalytic properties of the clicked MOFs were studied using the Knoevenagel condensation reactions between benzaldehyde and different methylene compounds. Only the NH2-functionalized MOF is active, suggesting that the amino group, rather than the triazole group or any other component of the framework, is the crucial active site. The catalysis is heterogeneous. The MOF is recyclable for the reaction with malononitrile but not for the reaction with ethyl cyanoacetate. The deactivation in the latter case is proposed to be because the amino site reacts with the ester group of ethyl cyanoacetate to form amide.
Introduction
Metal–organic frameworks (MOFs) have emerged as a new class of fascinating materials having potential applications in some fields such as gas adsorption/separation, catalysis, sensing and drug delivery.1–10 The modification of the pore surfaces with desired functional groups is important for achieving better performance in the above applications. Post-synthetic modification (PSM), which involves a reaction with any component of a known framework, is a very attractive strategy for modification of MOFs,11–13 but PSM is challenging because the modification may lead to undesired and unpredictable changes or destruction in the frameworks. The most widely studied platforms of covalent PSM are the MOFs bearing uncoordinated amino groups, which can readily react with aldehydes, isocyanates, and anhydrides so that various functional groups can be grafted onto the pore surfaces.14–16 More sophisticated reactions and stable MOFs suitable for PSM is still being sought not only to enrich the chemical diversity of MOFs but also to functionalize the materials with application-oriented groups.17–19The copper(I)-catalyzed azide–alkyne cycloaddition (CuAAC) reaction, a typical ‘click’ reaction, represents an elegant route for PSM of MOFs. A variety of alkyne-20–24 and azide-tagged25–28 MOFs have been successfully modified by CuAAC reactions. The metal-free strain-promoted azide–alkyne cycloaddition using cyclooctyne derivatives has also been employed for PSM of a mesoporous MOF.29 However, the alkyne- or azide-functionalized linkers and the stable MOFs applicable to click PSM are still quite limited in number.
The isoreticular Zr(IV) MOFs of general formula [Zr6O4(OH)4(L)6] [L = rigid (quasi-)linear dicarboxylates, such as 1,4-benzenedicarboxylate, biphenyl-4,4′-dicarboxylate and terphenyl-4,4′′-dicarboxylate for UiO-66, UiO-67 and UiO-68, respectively] contain face-sharing octahedral and tetrahedral cages with [Zr6(O)4(OH)4(COO)12] as vertices.30 The thermal and chemical stability of the Zr MOFs has allowed a number of recent studies related to absorption/separation,31–34 sensing,35,36 catalysis37–41 as well as PSM.42–48 However, only a few Zr–MOFs have been subjected to click PSM. Azidomethyl-functionalized UiO-68 MOFs were clicked with different alkynes to modify the pore surface for selective CO2 adsorption over N2 or to transform MOFs to polymer gels,49,50 and very recently, an azido-functionalized UiO-66 MOF (UiO-66–N3) has been clicked on the nanocrystal surfaces with alkyne-tagged oligonucleotides to create the first nucleic acid–MOF nanoparticle conjugates.51
In this article, we report the synthesis and the click PSM of a new Zr(II) MOF, UiO-67–N3, followed by a catalytic study with the modified MOFs (Scheme 1). UiO-67–N3 was synthesized from 2-azidobiphenyl-4,4′-dicarboxylic acid (H2BPDC–N3), a N3-functionalized linker not yet explored for the construction of MOFs. As will be shown, we have overcome the difficulty arising from the in situ thermocyclization of the ligand and successfully obtained the target N3-functionalized MOF. The PSM of UiO-67–N3 was performed via CuAAC click reactions with different alkynes [methyl propiolate (HC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CCOOMe), 3-butyn-1-ol (HCCCH2CH2OH) and propargylamine (HCCCH2NH2)] to produce new and more stable MOFs with different functionalities (UiO-67–Tz–COOCH3, UiO-67–Tz–OH and UiO-67–Tz–NH2). UiO-67–Tz–NH2 is a potential basic catalyst. The Knoevenagel condensation between carbonyl and activated methylene is well-known as a base-catalyzed C–C coupling reaction and has important applications in fine chemicals and pharmaceuticals. It has been demonstrated that the condensation can be catalyzed by different amino-functionalized MOFs, such as IRMOF-3, MIL-101–NH2, and UiO-66–NH2, with amino as active sites.52–56 Here we chose the reactions of benzaldehyde with ethyl cyanoacetate and malononitrile as model reactions to test the catalytic properties of the MOFs modified via click chemistry.
CCOOMe), 3-butyn-1-ol (HCCCH2CH2OH) and propargylamine (HCCCH2NH2)] to produce new and more stable MOFs with different functionalities (UiO-67–Tz–COOCH3, UiO-67–Tz–OH and UiO-67–Tz–NH2). UiO-67–Tz–NH2 is a potential basic catalyst. The Knoevenagel condensation between carbonyl and activated methylene is well-known as a base-catalyzed C–C coupling reaction and has important applications in fine chemicals and pharmaceuticals. It has been demonstrated that the condensation can be catalyzed by different amino-functionalized MOFs, such as IRMOF-3, MIL-101–NH2, and UiO-66–NH2, with amino as active sites.52–56 Here we chose the reactions of benzaldehyde with ethyl cyanoacetate and malononitrile as model reactions to test the catalytic properties of the MOFs modified via click chemistry.
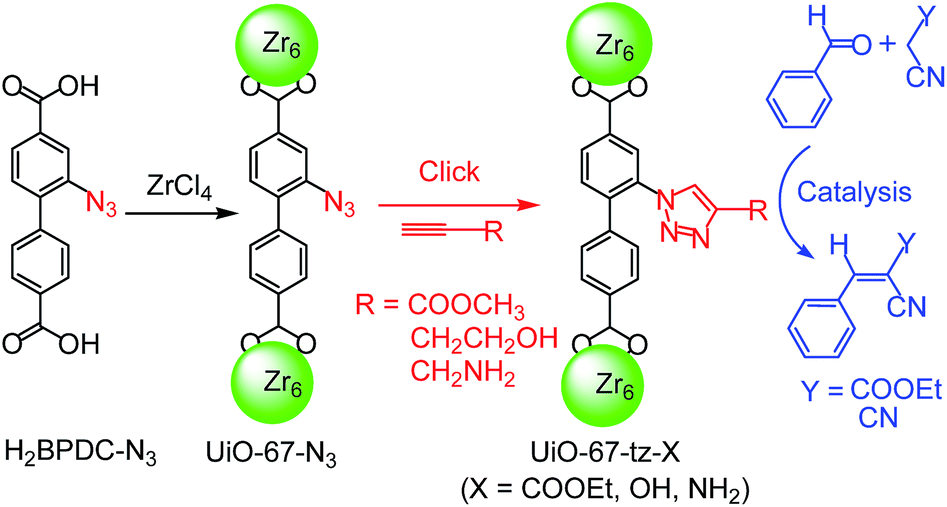 | ||
| Scheme 1 Synthesis and click PSM of UiO-67–N3 and the Knoevenagel condensation reactions studied. | ||
Experimental
Physical measurements
Elemental analyses were performed on an Elementar Vario ELIII analyzer. FT-IR spectra were recorded in the range of 500–4000 cm−1 using KBr pellets on a Nicolet NEXUS 670 spectrometer. Thermogravimetric Analysis (TGA) was performed on a Mettler TGALSDTA851e/5FL1100 instrument. Powder X-ray diffraction (XRD) measurements were performed on a Rigaku Ultima IV diffractometer equipped with Cu-Kα. Nitrogen adsorption–desorption measurements were performed at 77 K on a Quancachrome Autosorb-3B instrument after heating the samples at certain temperature for 6 h; the specific surface areas were calculated using the Brunauer–Emmett–Teller (BET) method. NMR spectra were recorded on a Bruker Avance 500 MHz NMR spectrometer. Gas chromatography (GC) analyses were performed on a LingHua GC 9890E instrument equipped with a Flame Ionization Detector (FID) detector.Synthesis of UiO-67–N3
All the solvents and reagents were commercially available and used as received.PSM of UiO-67–N3
The click PSM with the different alkynes caused distinct changes in colour, which are shown in Fig. S1.†
Catalytic studies
A given amount of as-synthesized UiO-67–Tz–NH2 (48 mg, 0.012 mmol. The amount of amino is about 0.07 mmol, i.e., 7 mol% with reference to benzaldehyde) was placed in a flask. After three cycles of vacuum pumping and nitrogen injection, DMF (3 mL) was added and the flask was kept at 40 °C in an oil bath. The methylene substrates (2 mmol) and benzaldehyde (1 mmol) was successively syringed to initiate the reaction, and the reaction was monitored by GC at different time intervals. For recyclability study, the solid after the catalytic reaction was isolated by centrifugation, washed with DMF several times, transferred to the reactor and used directly for the next run of catalytic reaction.Results and discussion
Synthesis and characterization of UiO-67–N3
The reaction temperature is crucially important for the synthesis of UiO-67–N3. Our initial synthetic attempt using the solvothermal conditions similar to that for UiO-67 was disappointing. The solvothermal reaction of ZrCl4 and H2BPDC–N3 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio) in DMF at 140 °C in the presence of acetic acid (HOAc) as modulator always led to an unidentified and essentially amorphous solid, according to XRD measurements (Fig. 1a). Worse still is that IR spectrum of the sample did not show the absorption band characteristic of the azide group (Fig. 2a). To probe the change of the ligand, the sample was treated with concentrated HCl(aq.), and the resulting solid was isolated for IR and 1H NMR studies (Fig. S2†). The 1H NMR spectrum of the solid dissolved in DMSO-d6 is in perfect agreement with 9H-carbazole-2,7-dicarboxylic acid (H2CDC). Consistently, the IR spectrum of the solid shows a characteristic ν(NH) sharp peak at ∼3345 cm−1 with no indication of νas(N3) absorption. So, it could be concluded that under above synthetic conditions the H2BPDC ligand underwent complete thermocyclization to give H2CDC (Scheme 2), which formed an unknown phase with Zr(II).
:1 molar ratio) in DMF at 140 °C in the presence of acetic acid (HOAc) as modulator always led to an unidentified and essentially amorphous solid, according to XRD measurements (Fig. 1a). Worse still is that IR spectrum of the sample did not show the absorption band characteristic of the azide group (Fig. 2a). To probe the change of the ligand, the sample was treated with concentrated HCl(aq.), and the resulting solid was isolated for IR and 1H NMR studies (Fig. S2†). The 1H NMR spectrum of the solid dissolved in DMSO-d6 is in perfect agreement with 9H-carbazole-2,7-dicarboxylic acid (H2CDC). Consistently, the IR spectrum of the solid shows a characteristic ν(NH) sharp peak at ∼3345 cm−1 with no indication of νas(N3) absorption. So, it could be concluded that under above synthetic conditions the H2BPDC ligand underwent complete thermocyclization to give H2CDC (Scheme 2), which formed an unknown phase with Zr(II).
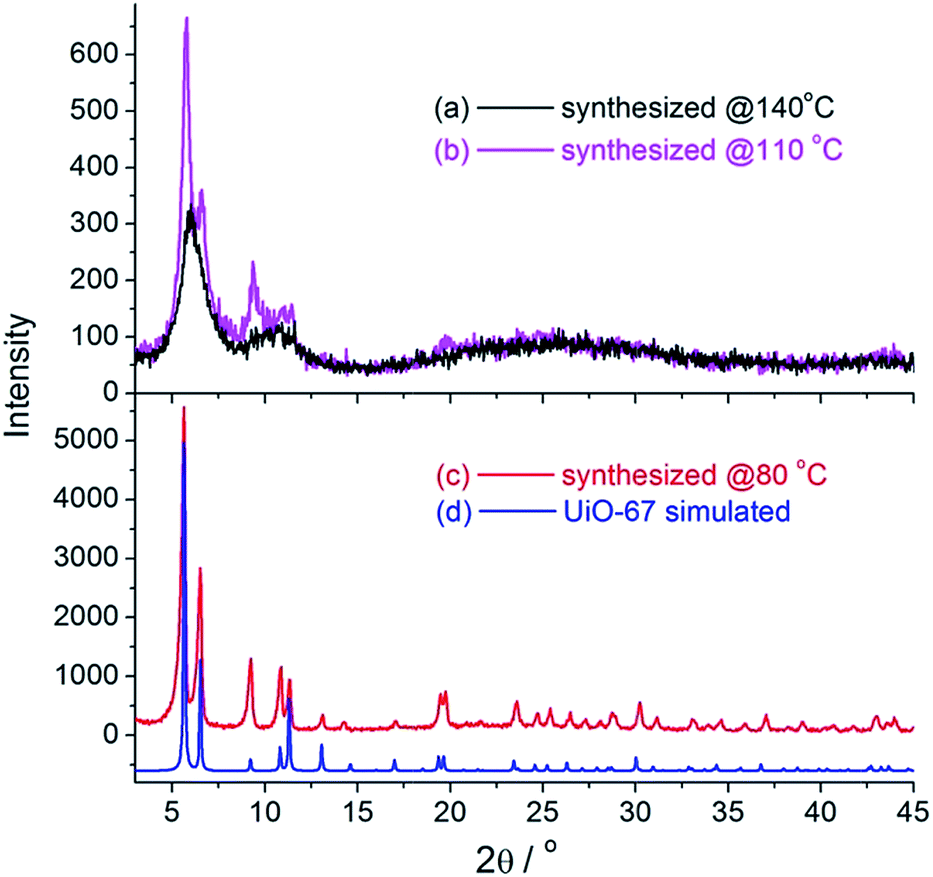 | ||
| Fig. 1 XRD patterns of the products synthesized at different temperature (a–c) and the simulated pattern for UiO-67 (d, with arbitrary intensity). Note that the reflection intensity of (c) is much high than that of (a and b). | ||
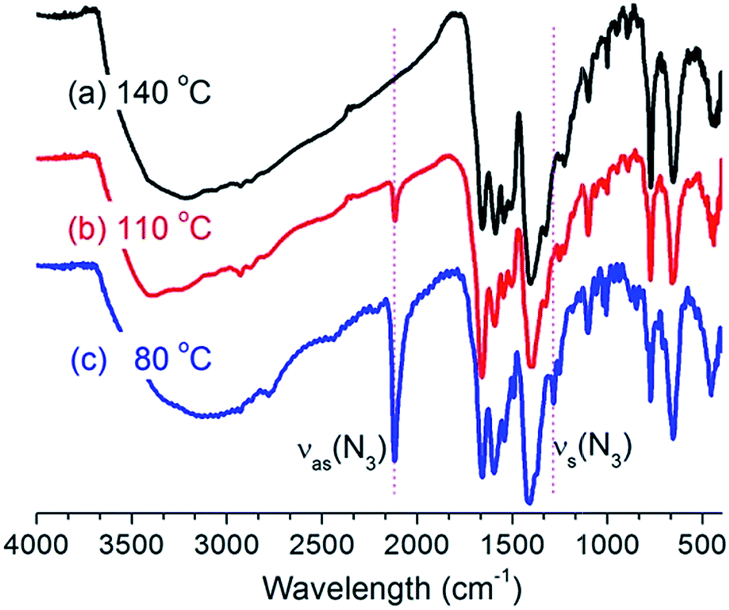 | ||
| Fig. 2 IR spectra of the products synthesized at different temperature. | ||
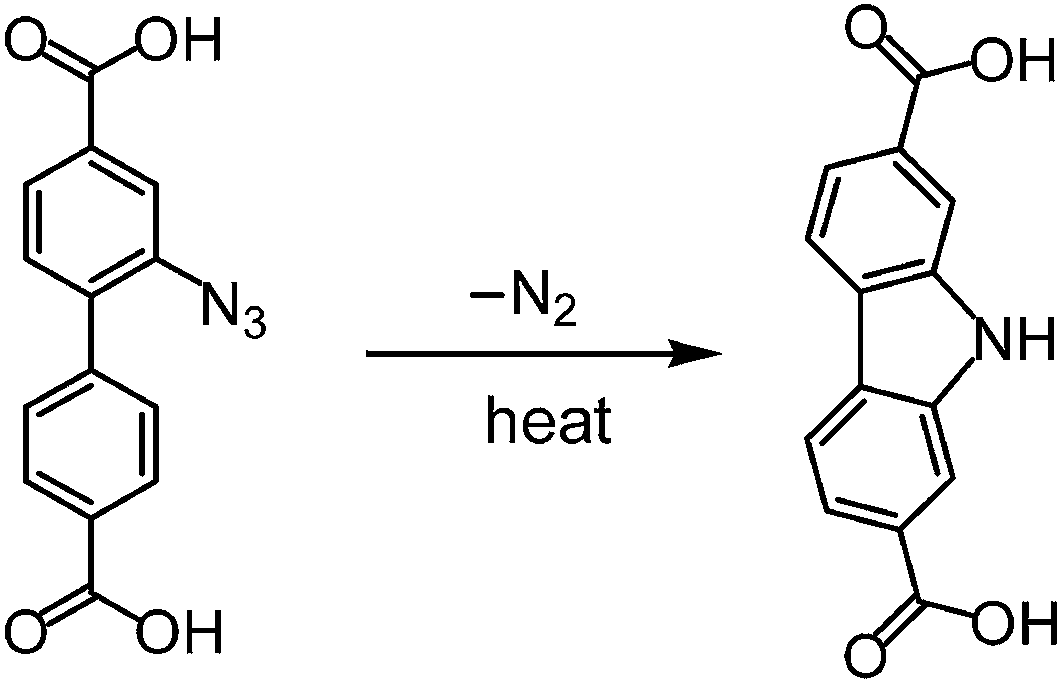 | ||
| Scheme 2 Thermocyclization of H2BPDC–N3 to H2CDC. | ||
To avoid the in situ ligand transformation, we sought to synthesize UiO-67–N3 at low temperature. The sample synthesized at 110 °C shows weak and sharp νas(N3) IR absorption at ∼2117 cm−1 (Fig. 2b), and the IR spectrum of the solid obtained by digesting the sample with concentrated HCl(aq.) shows both νas(N3) and ν(NH) bands (Fig. S3†), suggesting the coexistence of BPDC–N3 and CDC in the as-synthesized sample. Consistently, XRD data (Fig. 1. Compare (b) with (a) and (d)) indicates that a crystalline phase similar to UiO-67 was generated at 110 °C but commingled with some unknown amorphous phase(s) with the CDC ligand. The results encouraged us to further reduce the synthetic temperature to 80 °C, which gave good results. The IR spectrum of the product shows a very strong νas(N3) band at ∼2117 cm−1 together with a band at 1280 cm−1 attributable to νs(N3) (Fig. 2c). The 1H NMR and IR spectra after digestion show no evident signals of H2CDC and are identical to those of H2BPDC–N3 (Fig. S4†). These indicate that the thermocyclization decomposition of BPDC–N3 was prevented under the mild synthetic conditions. Furthermore, the XRD pattern of the product is well resolved with sharp and intense peaks and in good agreement with that of UiO-67 (Fig. 1c and d), with no evident indications of impure phases. So, UiO-67–N3 was successfully synthesized at 80 °C as a pure crystalline phase. The synthesis is reproducible.
The influence of auxiliary synthetic modulators, benzoic acid (HOBz) and HOAc, on the product was also investigated. In the absence of any modulating agent and under otherwise identical conditions, the reactions gave precipitates quickly. XRD indicated that the product isolated is also UiO-67–N3 (Fig. 3a) but the very broad reflections suggest that very small crystallites were obtained. The addition of HOBz into the reaction mixture slowed the precipitation and decreased the final yield. As can be see from Fig. 3b, the product obtained in the presence of 40 equivalents of HOBz shows less broad XRD peaks than that obtained in the absence of modulators, indicating that the slowed precipitation favours the crystal growth. Further increasing the amount of the modulator led to significant decrease in yield without apparent improvement in crystallinity. The reaction in the presence of 60 equivalents of HOBz did not yield any solid product within 2 weeks. The effect of HOAc on crystal growth is more pronounced. As shown in Fig. 3c, the product synthesized in the presence of 10 equivalents of HOAc shows very sharp and well-resolved reflections. The yield is moderate. No further optimization of the synthetic conditions was performed.
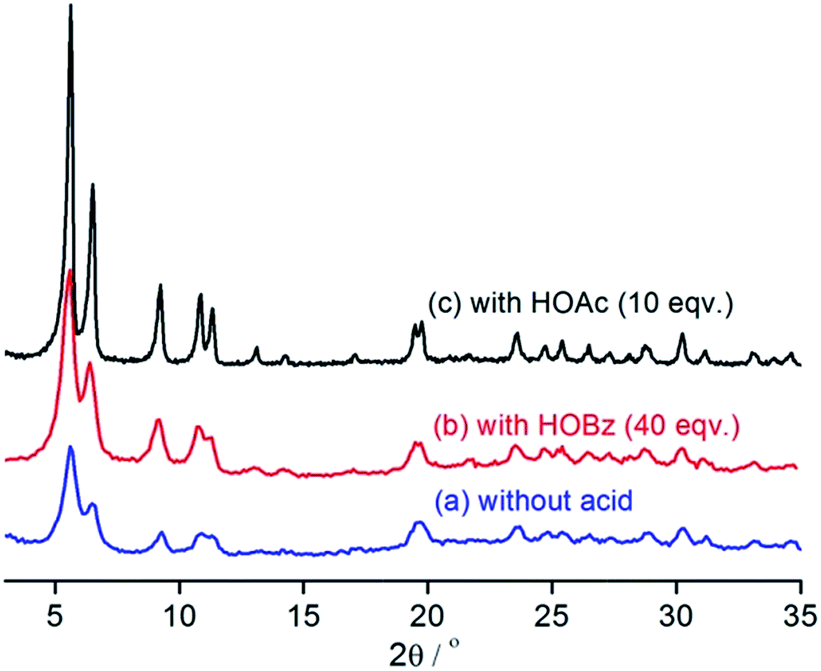 | ||
| Fig. 3 XRD patterns of the UiO-67–N3 samples synthesized in the absence/presence of modulators. | ||
The thermal behaviour of UiO-67–N3 was studied by TGA (Fig. S5†). UiO-67–N3 undergoes a rapid weight loss of ca. 43% when heated to 210 °C and a much slower loss upon further heating until another rapid loss appears above 440 °C. The weight loss above 440 °C corresponds to the complete decomposition of the framework, and the loss below 210 °C can be due to the release of the guest molecules (DMF and water) enclosed in the material and the N2 byproduct of the possible thermocyclization of BPDC–N3 (calculated total loss: 47%). The intermediate slow weight loss may be due to the dehydration of the [Zr6O4(OH)4] cluster and partial decomposition of the organic component. To probe the integrity of the framework during thermal treatments, the XRD patterns of the samples heated at different temperatures were measured (Fig. S6†). The broadening of the reflections is evident even at 60 °C, and only very broad reflections were observed for the sample heated at 110 °C, suggesting collapse of the framework. The 1H NMR spectrum of the sample heated at 110 °C and dissolved in a 20% NaOD/D2O solution indicates that most (∼80%) of the BPDC–N3 ligand has transformed into CDC (Fig. S7†). So the poor thermal stability of UiO-67–N3 is related to the thermocyclization of the BPDC–N3 ligand. The results also indicate that the resulting CDC ligand cannot support the framework. Furthermore, our attempts to synthesize a UiO-67-type framework from H2CDC as starting material have come out in vain, also implying that the UiO-67-type framework cannot be formed with CDC. Perhaps the bent shape of the CDC ligand (the orientations of the two coordinative carboxylate groups have a angle of about 150° (ref. 58)) disfavours the highly symmetric structure of UiO-67 type, which prefers a linear linkage between the 12-connected [Zr6O4(OH)4(COO)12] clusters.
The solvent resistance of UiO-67–N3 was also studied (Fig. S6†). Immersing the material in some common solvents including acetone, dichloromethane, trichloromethane and ethanol led to the loss of crystallinity, according to XRD measurements. N2 adsorption experiments revealed that the samples after solvent exchange and/or heating at 100 °C show only surface adsorption, also indicating collapse of the framework. The poor thermal and solvent resistance of UiO-67–N3 compared with UiO-67 reflect that the incorporation of a reactive group into a framework may cause dramatic decrease in stability, a challenging issue often encountered in preparing MOFs for PSM. Fortunately, however, the structural integrity of UiO-67–N3 remains intact in DMF: immersing it in DMF for a prolonged time did not lead to appreciable change in XRD. This made it possible to modify the MOF via the CuAAC click reaction, for which DMF is a frequently used solvent. It will be shown below that the thermal and chemical stabilities of the framework can be enhanced after the click modification, which further allows us to perform a catalytic study.
Post-synthetic modification by click chemistry
We have succeeded in post-synthetically transforming UiO-67–N3 into UiO-67–Tz–COOCH3, UiO-67–Tz–OH and UiO-67–Tz–NH2 via the CuAAC click reactions with methyl propiolate, 3-butyn-1-ol and propargylamine, respectively. The reactions were performed by stirring the mixture of UiO-67–N3, the alkynes and a catalytic amount of CuI in DMF at 60 °C. The reaction progress was monitored by IR spectra. A direct evidence for the occurrence of the click reaction is the change in the characteristic IR band of the azide group. As shown in Fig. 4 for the reaction with methyl propiolate, the νas(N3) band at ∼2117 cm−1 significantly decreased in intensity after 10 h and completely disappeared after 18 h, and so did the weaker νs(N3) band at ∼1280 cm−1. This clearly indicates the complete transformation of the azide group after 18 h. Meanwhile, a new band appeared at ∼1730 cm−1 in the product and grew with reaction time, which could be attributed to the carbonyl stretching band of the ester group. The absence of the ν(CC) absorption (∼2130 cm−1) indicates that the modified MOF does not contain unreacted methyl propiolate and that the ester group has been successfully introduced onto the MOF through the CuAAC click reaction between the alkyne group and the MOF.
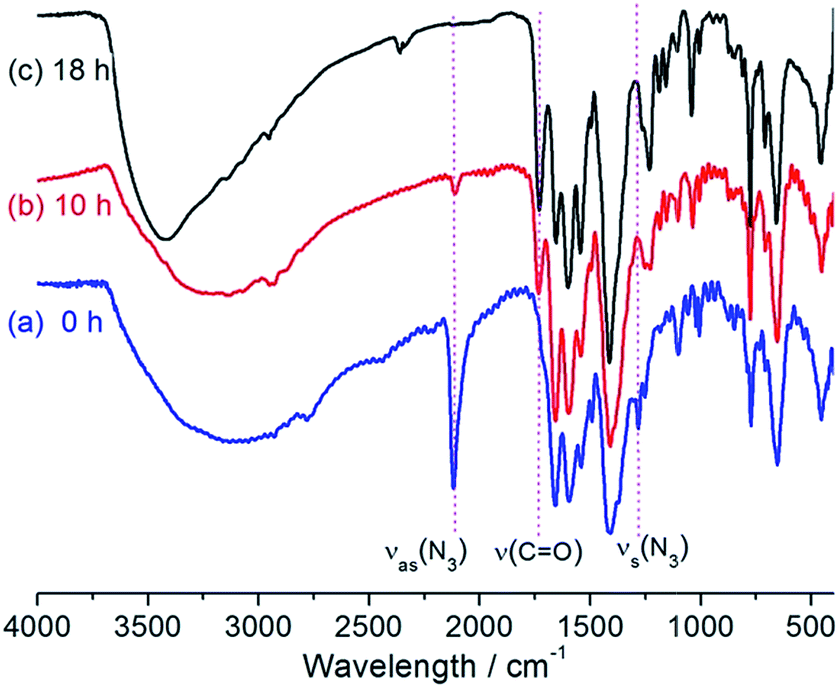 | ||
| Fig. 4 Changes in the IR spectra of UiO-67–N3 when reacted with methyl propiolate. | ||
The occurrence of the click reaction was further confirmed by the 1H NMR spectrum (Fig. 5) of the modified MOF (UiO-67–Tz–COOCH3) dissolved in HF(aq., 40%)/DMSO-d6 (v/v = 1/100), which is in perfect agreement with 2-(4-(methoxycarbonyl)-1,2,3-triazol-1-yl)biphenyl-4,4′-dicarboxylic acid, the product of the click reaction between H2BPDC–N3 and methyl propiolate. Furthermore, the XRD pattern of the MOF after the click reaction is almost identical to that of UiO-67–N3, indicating the retention of the framework integrity after the introduction of the 4-(methoxycarbonyl)-1,2,3-triazolyl group (Fig. 6).
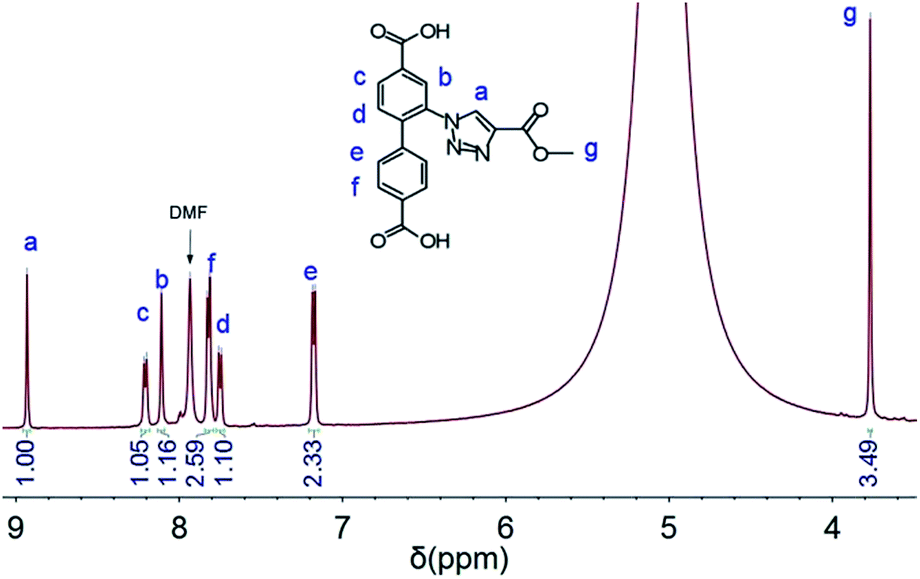 | ||
| Fig. 5 1H NMR spectrum of UiO-67–Tz–COOCH3 dissolved in HF(aq., 40%)/DMSO-d6. | ||
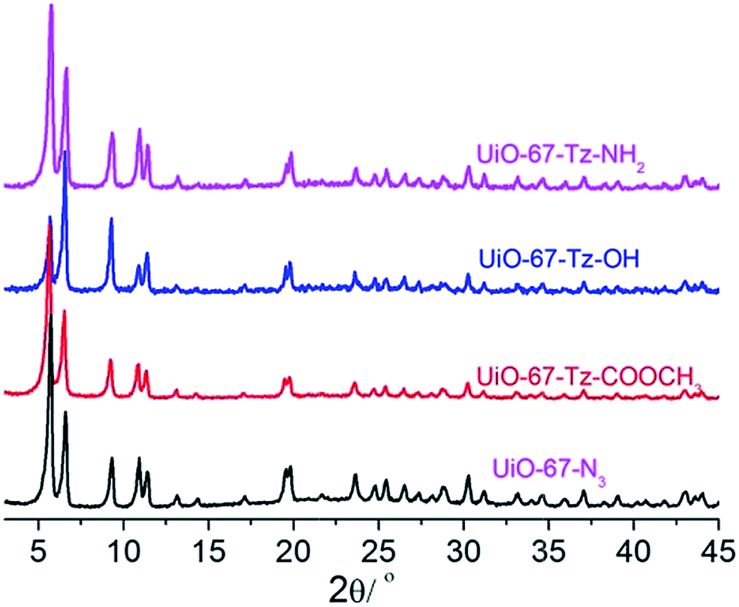 | ||
| Fig. 6 XRD of the modified MOFs compared with that of UiO-67–N3. | ||
The click PSM of UiO-67–N3 with 3-butyn-1-ol and propargylamine was also successful, giving UiO-67–Tz–OH and UiO-67–Tz–NH2, respectively, with the framework integrity retained. The XRD and spectral evidences are given in Fig. 6 and S8.†
Stability of the modified MOFs
Thermogravimetric measurements with the modified MOFs were performed under nitrogen atmosphere (Fig. S9†). They show more complex thermal behaviours than the azido-tagged mother materials, with multiple steps of weight loss, which are not easy to assign individually. The weight loss from room temperature to 400 °C is in the range of 29–35%, in fair agreement with the release of the guest molecules (DMF and water, calculated content: 32–33%) enclosed in the materials. Generally speaking, TGA data cannot give definite information about the thermal stability of the frameworks. Therefore, the stability was studied by means of XRD. Remarkably, the modified MOFs, especially UiO-67–Tz–NH2, show better thermal and chemical stabilities than the parent UiO-67–N3 MOF. XRD data showed that they are all stable in DMF. UiO-67–Tz–COOCH3 and UiO-67–Tz–OH can also stand heating at 90 °C and guest exchange with acetone and methanol (Fig. S10†), respectively, while UiO-67–N3 collapses when heated at 60 °C or immersed in these solvents. UiO-67–Tz–NH2 exhibits significantly higher stability. It can retain the framework integrity upon heating at 160 °C according to XRD measurements at various temperatures (Fig. 7). The high-angle reflections are weakened upon heating at higher temperature (140–160 °C), indicating the loss of short-range order to some degree, but the intensity can be recovered by immersing the sample in DMF or imposing the sample to moist air. UiO-67–Tz–NH2 also survives heating in DMF and solvent exchange with acetone and dichloromethane.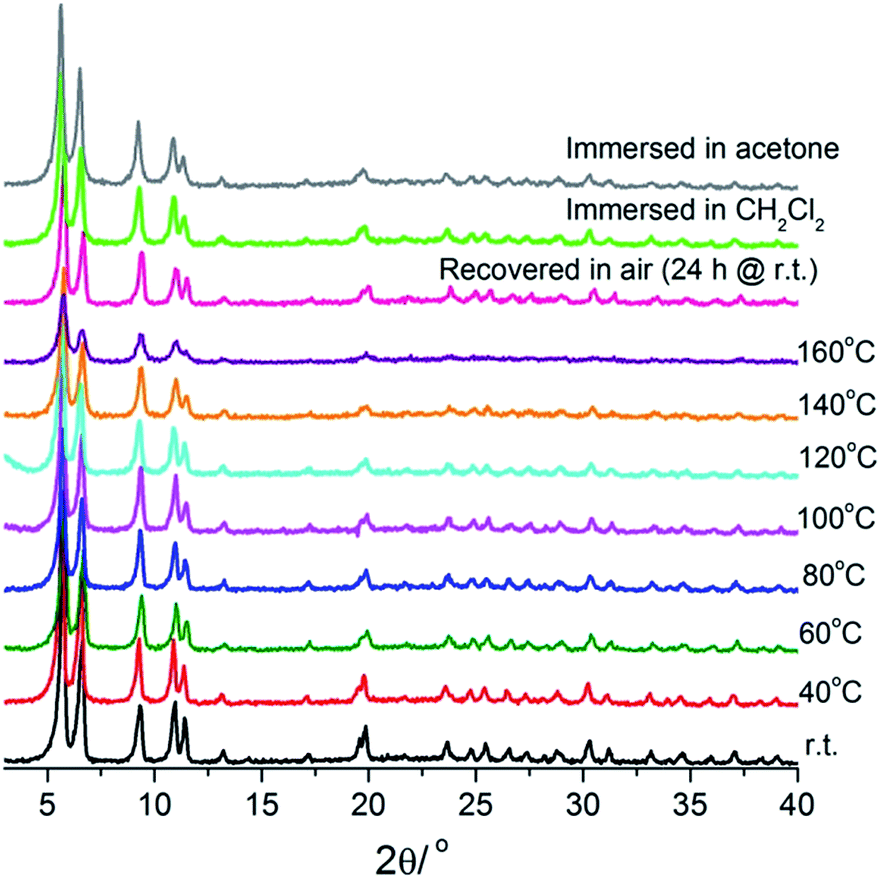 | ||
| Fig. 7 XRD patterns of UiO-67–Tz–NH2 heated at various temperature, recovered in moist air, and immersed in different solvents. | ||
The nitrogen adsorption–desorption isotherms of the modified MOFs are shown in Fig. S11.† Considering the thermal stability, UiO-67–Tz–COOCH3 and UiO-67–Tz–OH were heated at 90 °C in vacuum before the adsorption measurement. The BET (Langmuir) surface areas are 148 (211) and 84 (133) m2 g−1, respectively. The rather low surface areas are not surprising because the MOFs were not efficiently evacuated at 90 °C. UiO-67–Tz–NH2 shows a much larger BET (Langmuir) surface area of 404 (554) m2 g−1 after heating at 160 °C. Nevertheless, the area is much lower than that of UiO-67 (BET surface area > 2000 m2 g−1, depending upon the synthetic methods59,60). First, the incorporation of the triazole–CH2NH2 groups significantly reduces the pore size. Second, the framework may suffer some degree of local collapse upon heating at 160 °C, as indicated by XRD (see above).
Catalytic properties
To test the possible catalytic properties of the three MOFs modified via click chemistry, the Knoevenagel condensation reaction between benzaldehyde and ethyl cyanoacetate was performed. Considering that all of the MOFs we studied are stable in DMF and that DMF is a solvent widely used for Knoevenagel condensation,52,53,61 the catalytic tests were performed in DMF. DMF is also the solvent for preparing the MOFs. The MOFs isolated from the reaction mixture were washed with DMF and air-dried, and then used as catalysts without additional activation treatments.In the presence of UiO-67–Tz–NH2, the reaction between benzaldehyde and ethyl cyanoacetate (molar ratio, 1:2) proceeded smoothly at 40 °C to give ethyl (E)-α-cyanocinnamate as the only product. The conversion of benzaldehyde reached 86% after 2 h, and complete conversion was achieved within 8 h (Fig. 8). The catalytic reaction could be completed in 3 h if performed at 80 °C. For comparison, the blank control experiment (in the absence of any catalyst under otherwise identical conditions) gave a conversion of only 24% after 8 h at 40 °C. Evidently, UiO-67–Tz–NH2 serves as a catalyst for the Knoevenagel condensation. By contrast, UiO-67–Tz–OH and UiO-67–Tz–COOCH3 show no appreciable catalytic activity because the conversions over these two materials are comparable to that for the blank control experiment. It can be concluded that the catalytic activity of UiO-67–Tz–NH2 arises from the amino group, rather than the triazole group or the Zr–BPDC framework. It is generally accepted that the amino-catalyzed Knoevenagel condensation between benzaldehyde and ethyl cyanoacetate proceeds via an aldimine intermediate,52,62 which forms via a nucleophilic addition–elimination reaction between the aldehyde substrate and the amine group on the catalyst surface and reacts with the methylene substrate via another nucleophilic addition–elimination process to give the product and to recover the amino group.
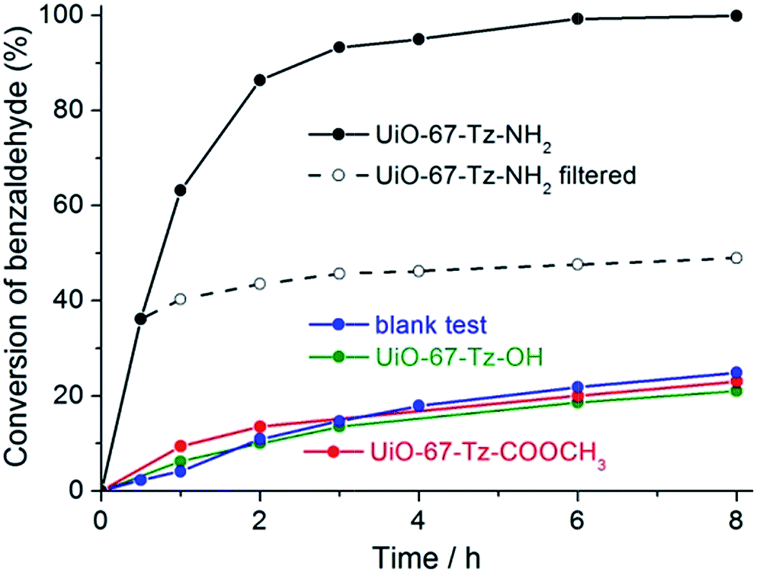 | ||
| Fig. 8 Conversion vs. time plots for Knoevenagel condensation between benzaldehyde (1 mmol) and ethyl cyanoacetate (2 mmol) in the presence or absence of different MOFs (0.012 mmol) in DMF (3 mL) at 40 °C. | ||
To clarify whether the catalytic activity arises from the solid or any homogeneous species leached in to the liquid phase, the catalyst was removed by hot filtration after reacting for 0.5 h, and the filtrate was kept under the same conditions and monitored by GC at different time intervals. As shown in Fig. 8, the reaction was almost stopped after filtering off the solid. The very slight increase in conversion could be due to a thermally activated slow reaction, as occurred in the blank test. The observation indicates that the catalysis over UiO-67–Tz–NH2 is heterogeneous, the catalytic site arising from the solid phase.
The XRD pattern of the used catalyst is in a good agreement with that of the fresh catalyst (Fig. S12†), indicating that the crystalline framework remains intact after the reaction. However, the used catalyst became completely inactive when reused for the second run (the conversion of benzaldehyde is only 27% after 8 h at 40 °C, similar to that for a blank test). We assumed that the deactivation could because the active aliphatic amino group reacted with the ester group of ethyl cyanoacetate to give amide, which is too weak in basicity to catalyze the reaction. The amide formation is supported by a comparison between the FTIR spectra of the fresh and used catalysts (Fig. S12†). For the used catalyst, the weak peak at 2225 cm−1 can be attributed to the cyano group attached to the framework through amide formation, and the additional shoulder absorption at about 1700 cm−1 is attributable to the carbonyl vibration of the amide group (the carbonyl vibration of ethyl cyanoacetate appears at 1760 cm−1). The results suggest that UiO-67–Tz–NH2 can be further modified without sacrificing the integrity of the crystalline framework, which may be a subject of further studies.
To further demonstrate the catalytic activity of UiO-67–Tz–NH2 and to avoid the above deactivation mechanism, benzaldehyde was reacted with malononitrile, an active methylene compound having no ester group. It turned out that the reaction over UiO-67–Tz–NH2 reached completion within 3 h (Fig. S13†), much more rapid than the blank test in absence of catalysts. The fact that malononitrile reacted with benzaldehyde more rapidly than ethyl cyanoacetate under identical catalytic conditions is consistent with the different intrinsic reactivity (acidity) of the methylene compounds.8,53 Interestingly, for the reaction between benzaldehyde with malononitrile, the second and third runs using the recovered catalyst showed no significant decrease in conversion (Fig. 9). The slight decrease can be simply due to the loss of the catalyst during the recycling. The results support the proposal that the deactivation for the reaction with ethyl cyanoacetate is related to the ester group.
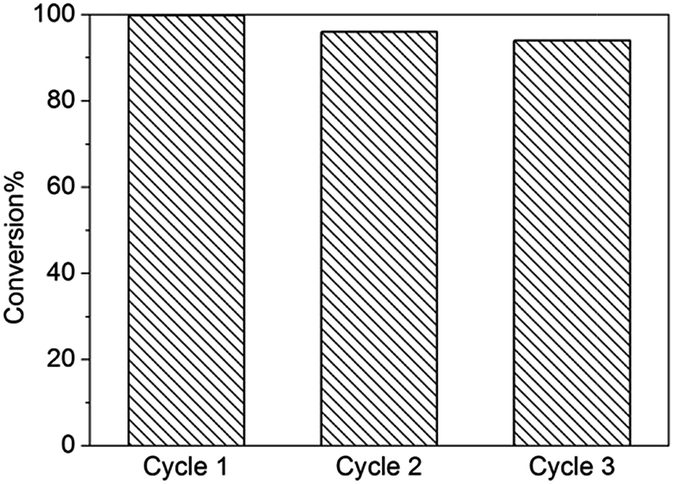 | ||
| Fig. 9 Conversion data of the Knoevenagel condensation reactions (3 h at 40 °C) between benzaldehyde and malononitrile over fresh and recovered UiO-67–Tz–NH2 catalyst. | ||
Conclusions
A new azido-functionalized Zr(II) MOF, UiO-67–N3, was successfully synthesized at relatively low temperature, which avoids the in situ thermocyclization of the H2BPDC–N3 ligand. The PSM of UiO-67–N3 was successfully performed via CuAAC click reactions with alkyne compounds bearing different functionalities. The clicked MOFs, especially UiO-67–Tz–NH2, exhibit better stabilities than the mother material UiO-67–N3. UiO-67–Tz–NH2 is a heterogeneous catalyst for Knoevenagel condensation, the activity arising from the amino group rather than the triazole group or the framework. The MOF is recyclable for the reaction with malononitrile but not for ethyl cyanoacetate because the ester group of the latter substrate reacts with and hence deactivate the amino site. This work demonstrates the efficiency of the click-chemistry approach of PSM and the potentials in engineering MOF catalysts by PSM, and it also illustrates the cases in which one needs to get a balance between stability and reactivity when choosing organic linkers and metal–organic frameworks for PSM.Acknowledgements
This work is supported by the National Natural Science Foundation of China (NSFC no. 21173083) and the Research Fund for the Doctoral Program of Higher Education of China.Notes and references
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- C. Wang, D. Liu and W. Lin, J. Am. Chem. Soc., 2013, 135, 13222–13234 CrossRef CAS PubMed.
- W. Xuan, C. Zhu, Y. Liu and Y. Cui, Chem. Soc. Rev., 2012, 41, 1677–1695 RSC.
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T. H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724–781 CrossRef CAS PubMed.
- J. R. Li, J. Sculley and H. C. Zhou, Chem. Rev., 2012, 112, 869–932 CrossRef CAS PubMed.
- P. Horcajada, R. Gref, T. Baati, P. K. Allan, G. Maurin, P. Couvreur, G. Férey, R. E. Morris and C. Serre, Chem. Rev., 2012, 112, 1232–1268 CrossRef CAS PubMed.
- L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. Van Duyne and J. T. Hupp, Chem. Rev., 2012, 112, 1105–1125 CrossRef CAS PubMed.
- A. Corma, H. García and F. X. Llabres i Xamena, Chem. Rev., 2010, 110, 4606–4655 CrossRef CAS PubMed.
- A. Dhakshinamoorthy, M. Alvaro and H. García, Chem. Commun., 2012, 48, 11275–11288 RSC.
- A. Dhakshinamoorthy, M. Opanasenko, J. Cejka and H. García, Catal. Sci. Technol., 2013, 3, 2509–2540 CAS.
- Z. Wang and S. M. Cohen, Chem. Soc. Rev., 2009, 38, 1315–1329 RSC.
- S. M. Cohen, Chem. Rev., 2012, 112, 970–1000 CrossRef CAS PubMed.
- K. Manna, T. Zhang and W. Lin, J. Am. Chem. Soc., 2014, 136, 6566–6569 CrossRef CAS PubMed.
- D. J. Lun, G. I. N. Waterhouse and S. G. Telfer, J. Am. Chem. Soc., 2011, 133, 5806–5809 CrossRef CAS PubMed.
- C. Volkringer and S. M. Cohen, Angew. Chem., Int. Ed., 2010, 49, 4644–4648 CrossRef CAS PubMed.
- J. Chen, R. Liu, H. Gao, L. Chen and D. Ye, J. Mater. Chem. A, 2014, 2, 7205–7213 CAS.
- C. Chen, C. A. Allen and S. M. Cohen, Inorg. Chem., 2011, 50, 10534–10536 CrossRef CAS PubMed.
- J. Juan-Alcañiz, J. Ferrando-Soria, I. Luz, P. Serra-Crespo, E. Skupien, V. P. Santos, E. Pardo, F. X. Llabres i Xamena, F. Kapteijn and J. Gascon, J. Catal., 2013, 307, 295–304 CrossRef PubMed.
- D. Jiang, L. L. Keenan, A. D. Burrows and K. J. Edler, Chem. Commun., 2012, 48, 12053–12055 RSC.
- P. Deria, W. Bury, J. T. Hupp and O. K. Farha, Chem. Commun., 2014, 50, 1965–1968 RSC.
- T. Gadzikwa, O. K. Farha, C. D. Malliakas, M. G. Kanatzidis, J. T. Hupp and S. T. Nguyen, J. Am. Chem. Soc., 2009, 131, 13613–13615 CrossRef CAS PubMed.
- D. Zhao, S. Tan, D. Yuan, W. Lu, Y. H. Rezenom, H. Jiang, L.-Q. Wang and H.-C. Zhou, Adv. Mater., 2011, 23, 90–93 CrossRef CAS PubMed.
- W. Zhu, C. He, P. Wu, X. Wu and C. Duan, Dalton Trans., 2012, 41, 3072–3077 RSC.
- P. Roy, A. Schaate, P. Behrens and A. Godt, Chem.–Eur. J., 2012, 18, 6979–6985 CrossRef CAS PubMed.
- Y. Goto, H. Sato, S. Shinkai and K. Sada, J. Am. Chem. Soc., 2008, 130, 14354–14355 CrossRef CAS PubMed.
- M. Savonnet, D. Bazer-Bachi, N. Bats, J. Perez-Pellitero, E. Jeanneau, V. Lecocq, C. Pinel and D. Farrusseng, J. Am. Chem. Soc., 2010, 132, 4518–4519 CrossRef CAS PubMed.
- M. Savonnet, A. Camarata, J. Canivet, D. Bazer-Bachi, N. Bats, V. Lecocq, C. Pinel and D. Farrusseng, Dalton Trans., 2012, 41, 3945–3948 RSC.
- G. Tuci, A. Rossin, X. Xu, M. Ranocchiari, J. A. van Bokhoven, L. Luconi, I. Manet, M. Melucci and G. Giambastiani, Chem. Mater., 2013, 25, 2297–2308 CrossRef CAS.
- C. Liu, T. Li and N. L. Rosi, J. Am. Chem. Soc., 2012, 134, 18886–18888 CrossRef CAS PubMed.
- C. J. Hafizovic, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850–13851 CrossRef PubMed.
- Q. Yang, V. Guillerm, F. Ragon, A. D. Wiersum, P. L. Llewellyn, C. Zhong, T. Devic, C. Serre and G. Maurin, Chem. Commun., 2012, 48, 9831–9833 RSC.
- S. Biswas, J. Zhang, Z. Li, Y.-Y. Liu, M. Grzywa, L. Sun, D. Volkmer and D. V. P. Van, Dalton Trans., 2013, 42, 4730–4737 RSC.
- D. Cunha, C. Gaudin, I. Colinet, P. Horcajada, G. Maurin and C. Serre, J. Mater. Chem. B, 2013, 1, 1101–1108 RSC.
- D. H. Hong and M. P. Suh, Chem.–Eur. J., 2014, 20, 426–434 CrossRef CAS PubMed.
- K.-K. Yee, N. Reimer, J. Liu, S.-Y. Cheng, S.-M. Yiu, J. Weber, N. Stock and Z. Xu, J. Am. Chem. Soc., 2013, 135, 7795–7798 CrossRef CAS PubMed.
- J. Aguilera-Sigalat and D. Bradshaw, Chem. Commun., 2014, 50, 4711–4713 RSC.
- F. Vermoortele, M. Vandichel, B. Van de Voorde, R. Ameloot, M. Waroquier, V. Van Speybroeck and D. E. De Vos, Angew. Chem., Int. Ed., 2012, 51, 4887–4890 CrossRef CAS PubMed.
- J. Juan-Alcañiz, R. Gielisse, A. B. Lago, E. V. Ramos-Fernandez, P. Serra-Crespo, T. Devic, N. Guillou, C. Serre, F. Kapteijn and J. Gascon, Catal. Sci. Technol., 2013, 3, 2311–2318 Search PubMed.
- T. Toyao, M. Saito, Y. Horiuchi and M. Matsuoka, Catal. Sci. Technol., 2014, 4, 625–628 CAS.
- D. Sun, Y. Fu, W. Liu, L. Ye, D. Wang, L. Yang, X. Fu and Z. Li, Chem.–Eur. J., 2013, 19, 14279–14285 CrossRef CAS PubMed.
- F. Vermoortele, B. Bueken, B. G. Le, B. Van de Voorde, M. Vandichel, K. Houthoofd, A. Vimont, M. Daturi, M. Waroquier, V. Van Speybroeck, C. Kirschhock and D. E. De Vos, J. Am. Chem. Soc., 2013, 135, 11465–11468 CrossRef CAS PubMed.
- M. Kim, J. A. Boissonnault, P. V. Dau and S. M. Cohen, Angew. Chem., Int. Ed., 2011, 50, 12193–12196 CrossRef CAS PubMed.
- S. J. Garibay and S. M. Cohen, Chem. Commun., 2010, 46, 7700–7702 RSC.
- M. Pintado-Sierra, A. M. Rasero-Almansa, A. Corma, M. Iglesias and F. Sanchez, J. Catal., 2013, 299, 137–145 CrossRef CAS PubMed.
- M. Kandiah, S. Usseglio, S. Svelle, U. Olsbye, K. P. Lillerud and M. Tilset, J. Mater. Chem., 2010, 20, 9848–9851 RSC.
- M. Kim, J. F. Cahill, H. Fei, K. A. Prather and S. M. Cohen, J. Am. Chem. Soc., 2012, 134, 18082–18088 CrossRef CAS PubMed.
- P. W. Siu, Z. J. Brown, O. K. Farha, J. T. Hupp and K. A. Scheidt, Chem. Commun., 2013, 49, 10920–10922 RSC.
- H. Fei, J. Shin, Y. S. Meng, M. Adelhardt, J. Sutter, K. Meyer and S. M. Cohen, J. Am. Chem. Soc., 2014, 136, 4965–4973 CrossRef CAS PubMed.
- H.-L. Jiang, D. Feng, T.-F. Liu, J.-R. Li and H.-C. Zhou, J. Am. Chem. Soc., 2012, 134, 14690–14693 CrossRef CAS PubMed.
- T. Ishiwata, Y. Furukawa, K. Sugikawa, K. Kokado and K. Sada, J. Am. Chem. Soc., 2013, 135, 5427–5432 CrossRef CAS PubMed.
- W. Morris, W. E. Briley, E. Auyeung, M. D. Cabezas and C. A. Mirkin, J. Am. Chem. Soc., 2014, 136, 7261–7264 CrossRef CAS PubMed.
- J. Gascon, U. Aktay, M. D. Hernandez-Alonso, G. P. M. van Klink and F. Kapteijn, J. Catal., 2009, 261, 75–87 CrossRef CAS PubMed.
- Y. Yang, H.-F. Yao, F.-G. Xi and E.-Q. Gao, J. Mol. Catal. A: Chem., 2014, 390, 198–205 CrossRef CAS PubMed.
- M. Hartmann and M. Fischer, Microporous Mesoporous Mater., 2012, 164, 38–43 CrossRef CAS PubMed.
- F. X. Llabres i Xamena, F. G. Cirujano and A. Corma, Microporous Mesoporous Mater., 2012, 157, 112–117 CrossRef CAS PubMed.
- P. Wu, J. Wang, Y. Li, C. He, Z. Xie and C. Duan, Adv. Funct. Mater., 2011, 21, 2788–2794 CrossRef CAS.
- V. K. Olkhovik, D. A. Vasilevskii, A. A. Pap, G. V. Kalechyts, Y. V. Matveienko, A. G. Baran, N. A. Halinouski and V. G. Petushok, ARKIVOC, 2008, 9, 69–93 CrossRef.
- X.-C. Yi, M.-X. Huang, Y. Qi and E.-Q. Gao, Dalton Trans., 2014, 43, 3691–3697 RSC.
- A. Schaate, P. Roy, A. Godt, J. Lippke, F. Waltz, M. Wiebcke and P. Behrens, Chem.–Eur. J., 2011, 17, 6643–6651 CrossRef CAS PubMed.
- M. J. Katz, Z. J. Brown, Y. J. Colon, P. W. Siu, K. A. Scheidt, R. Q. Snurr, J. T. Hupp and O. K. Farha, Chem. Commun., 2013, 49, 9449–9451 RSC.
- M. J. Climent, A. Corma, I. Domínguez, S. Iborra, M. J. Sabater and G. Sastre, J. Catal., 2007, 246, 136–146 CrossRef CAS PubMed.
- R. Cortese and D. Duca, Phys. Chem. Chem. Phys., 2011, 13, 15995–16004 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra09883h |
| This journal is © The Royal Society of Chemistry 2015 |