
Plasmonic cooperation effect of metal nanomaterials at Au–TiO2–Ag interface to enhance photovoltaic performance for dye-sensitized solar cells†
Abstract
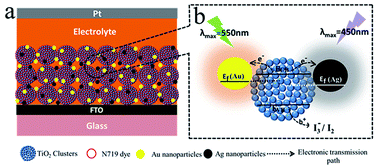
From the plasmonic cooperation effect of metal nanomaterials at a Au–TiO2–Ag interface, Au and Ag used complementary light-harvesting to enhance photovoltaic performance in dye-sensitized solar cells (DSSC). The best efficiency (η) of DSSC reached 7.51%, compared with 6.23% for pure TiO2 electrode. The average energy conversion efficiency and photocurrent density were increased by 20.8% and 29.9% compared with those of pure TiO2 electrodes. Hence, the complementary light-harvesting using different light absorption positions of Au and Ag nanomaterials and plasmonic cooperation effect of Au and Ag together improved the light harvesting, short circuit current density, open circuit voltage and photo-electric conversion efficiency in DSSCs.
 Please wait while we load your content...
Please wait while we load your content...

