
First-principles study of negative thermal expansion mechanism in A-site-ordered perovskite SrCu3Fe4O12
Abstract
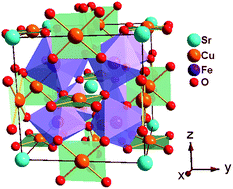
Negative thermal expansion (NTE) materials offer a tremendous opportunity for fundamental as well as applied research, yet the origin remains difficult to understand. Here we perform a systematic first-principles calculation to investigate electrical and magnetic properties of an A-site-ordered perovskite SrCu3Fe4O12 and clarify its NTE mechanism. We find that SrCu3Fe4O12 is an antiferromagnetic metal, and its magnetic ordering can be demonstrated as C-type within Fe ions of mixed valence at the B-site and paramagnetic Cu3+ at the A-site. Electronic structure analysis reveals that there occurs temperature-induced Cu–Fe intersite charge transfer, which is mediated by the corner-sharing O atoms. Meanwhile, the magnetic interaction is found to undergo a transition from antiferromagnetism to ferrimagnetism. Further phonon calculations demonstrate that the SrCu3Fe4O12 is thermodynamically stable both at low and high temperature, and that there appear degenerated phonon branches, indicating that it is easy to transfer energy between these modes. We also find that the large NTE of SrCu3Fe4O12 originates from intermetallic charge transfer induced by temperature, which relaxes the Sr–O and Fe–O bonding units in the oxide.
 Please wait while we load your content...
Please wait while we load your content...

