
Concentration dependent speciation and mass transport properties of switchable polarity solvents
Abstract
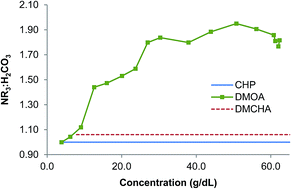
Tertiary amine switchable polarity solvents (SPS) consisting of predominantly water, tertiary amine, and tertiary ammonium and bicarbonate ions were produced at various concentrations for three different amines: N,N-dimethylcyclohexylamine, N,N-dimethyloctylamine, and 1-cyclohexylpiperidine. These amines exhibit either osmotic or non-osmotic character as observed through forward osmosis, which led to this study to better understand speciation and its influence on water transport through a semi-permeable membrane. For all concentrations, several physical properties were measured including viscosity, molecular diffusion coefficients, freezing point depression, and density. Based on these measurements, a variation on the Mark–Houwink equation was developed to predict the viscosity of any tertiary amine SPS as a function of concentration using the amine's molecular mass. The physical properties of osmotic SPS, which are identified as having an amine to carbonic acid salt ratio of ∼1 : 1, have consistent concentration dependence behavior over a wide range of concentrations, which suggests osmotic pressures based on low concentrations freezing point studies can be extrapolated reliably to higher concentrations. The observed physical properties also allowed the identification of solution state speciation of non-osmotic SPS, where the amine to carbonic acid salts ratio is significantly greater than one. These results indicate that, at most concentrations, the stoichiometric excess of amine is involved in solvating a proton with two amines.
 Please wait while we load your content...
Please wait while we load your content...

