
Controlling the microstructure of MFI zeolites with Mg(OH)2 nanocrystals to improve their catalytic performances†
Huijuan Weia,
Ning Zhanga,
Tian Zhaoa,
Yangqing Liua,
Yiqiang Wena,
Xiangyu Wang*a and
Baojun Li*ab
aInstitute of Industrial Catalysis, College of Chemistry and Molecular Engineering, Zhengzhou University, 100 Science Road, Zhengzhou 450001, P R China. E-mail: xiangyuwang@zzu.edu.cn; lbjfcl@zzu.edu.cn
bDepartment of Chemistry, Tsinghua University, Beijing 100084, P R China
First published on 3rd December 2014
Abstract
Control of the microstructure and morphology of molecular sieve crystals will significantly affect their catalytic performances. In this paper, titanium silicalite-1 (TS-1) and silicalite-1 (S-1) crystals were synthesized under hydrothermal conditions in the presence of Mg(OH)2 nanocrystals (NCs). XRD, TEM, FT-IR, and N2 adsorption were used to characterize the structure of TS-1 and S-1 crystals. The introduction of Mg(OH)2 NCs endow the as-synthesized TS-1 and S-1 with a much more rugged surface and some voids and slits via attachment onto the surface or entrapment inside TS-1 and S-1 crystals. The crystallinity behavior, microstructure and morphology of TS-1 and S-1 also are effectively affected and modified by the presence of Mg(OH)2 NCs. The obtained TS-1 and S-1 crystals were examined as catalysts for the cyclohexanone ammoximation and vapor phase Beckmann rearrangement of cyclohexanone oxime, respectively. The highest catalytic performances of the two reactions had been obtained when the Mg(OH)2 additive amount was 10 wt%. The catalytic performance was improved when the appropriate amount of Mg(OH)2 NCs was introduced into the MFI zeolite synthesis solution.
1. Introduction
The morphology and microstructure of molecular sieves often have significant effect on their sorption and catalytic performances.1–3 Synthesis condition adjustments,4 micro emulsion synthesis,5–7 template assisted methods,8–10 post-treatment11,12 and self-assembly13 have been extensively used to alter the morphologies and microstructures of the molecular sieves. Some micelles, carbon particles, aero gels, surfactants and polymers also played important role as templates for the construction of hierarchical porous molecular sieves.14–18 A steam-assisted conversion procedure has been adopted to self-assemble hierarchical porous beta zeolite.19 Titanium silicalite-1 crystals were self-assembled by controlled evaporation of colloidal suspensions.20 These approaches have not been widely extended to the synthesis of other molecular sieves. A cheap recyclable template assisted procedure and other simple strategies are needed for the industrial application of morphology and microstructure controllable molecular sieves.Over the last decade, inorganic nanocrystals (NCs) have attracted tremendous interest due to their tunable morphologies, structure and size-dependent properties.21 Some inorganic NCs were employed as hard templates to modify the crystallization behavior and microstructure of molecular sieve.22 Due to interfacial interaction, inorganic NCs can be entrapped into molecular sieves during the crystallization. After removal of inorganic NCs, hierarchical porous molecular sieve were achieved.
ε-Caprolactam is the main raw for the production of Nylon-6.23 Strenuous efforts have been devoted to developing environment-friendly new synthesis processes to avoid the several disadvantages in current industrial route such as reactor corrosion, environmental pollutions and by-production of a large amount of ammonium sulphate.24–26 The new synthesis process including the ammoximation reaction of cyclohexanone and the Beckmann rearrangement of cyclohexanone oxime has attracted widespread interest due to most potential industrial prospect (seen in Scheme 1).27 TS-1 and S-1 with MFI topology has always been reported to be the most highly active and selective catalysts for the cyclohexanone ammoximation and vapor phase Beckmann rearrangement of cyclohexanone oxime, respectively.28–31 This predicament drives us to apply environmental benign inorganic NCs into the synthesis process of TS-1 and S-1 molecular sieves, carry out the cyclohexanone ammoximation in TS-1 and the vapor phase Beckmann rearrangement of cyclohexanone oxime in S-1, finally research the relation between the microstructure and catalytic performance of zeolites.
 | ||
| Scheme 1 The reaction routes for cyclohexanone ammoxidation and Beckmann rearrangement of cyclohexanoe oxime. | ||
In this paper, TS-1 and S-1 were synthesized by introducing and removing Mg(OH)2 NCs into synthesis system to adjust the morphologies and microstructures of TS-1 and S-1 (seen in Scheme 2). The as-synthesized TS-1 and S-1 exhibited much improved catalytic performance in ammoximation of cyclohexanone and vapor phase Beckmann rearrangement of cyclohexanone oxime, respectively.
 | ||
| Scheme 2 Synthesis process of TS-1 and S-1 in the presence of Mg(OH)2 NCs. | ||
2. Experimental section
2.1. Synthesis
Mg(OH)2 nanocrystals (NCs) were prepared according to the literature.32 In a typical TS-1 synthesis, a certain amount of TPABr, aqueous ammonia and Mg(OH)2 NCs were added to colloidal silica at room temperature under stirring, the mixture was stirred for 0.5 h, and then TiCl3 was added dropwise. The molar composition of the gel was SiO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.02TiO2:0.10TPABr:nMg(OH)2:5NH3:30H2O. TS-1 with different n ratios of 0, 0.052, 0.10, 0.31 and 0.52 were synthesized. The corresponding mass percentage in total mass SiO2 was 0, 5, 10, 30 and 50 wt%, respectively. TS-1 particles of approximately 300 nm in size were used as seeds. The resulting gel was stirred for 24 h, then transferred to a Teflon lined autoclave and crystallized for 96 h at 175 °C. The obtained solid was washed with deionized water, dried at 120 °C overnight. The obtained composite was treated with HCl and H2O2 at 80 °C for 2 h for three times to remove the Mg(OH)2 NCs and non-framework titanium species.
:0.02TiO2:0.10TPABr:nMg(OH)2:5NH3:30H2O. TS-1 with different n ratios of 0, 0.052, 0.10, 0.31 and 0.52 were synthesized. The corresponding mass percentage in total mass SiO2 was 0, 5, 10, 30 and 50 wt%, respectively. TS-1 particles of approximately 300 nm in size were used as seeds. The resulting gel was stirred for 24 h, then transferred to a Teflon lined autoclave and crystallized for 96 h at 175 °C. The obtained solid was washed with deionized water, dried at 120 °C overnight. The obtained composite was treated with HCl and H2O2 at 80 °C for 2 h for three times to remove the Mg(OH)2 NCs and non-framework titanium species.
For S-1 synthesis, an aqueous suspension of Mg(OH)2 NCs was added into aqueous solution of TPAOH. The mixture was stirred for 30 min, followed by addition of TEOS. The molar compositions of synthesis mixtures were as follows: TEOS:0.20 TPAOH:nMg(OH)2:20H2O. S-1 with different n ratios of 0, 0.052, 0.10, 0.16 and 0.26 were synthesized. The corresponding mass percentage in total mass SiO2 was 0, 5, 10, 15 and 25 wt%, respectively. The resulting mixture was stirred vigorously at room temperature for 3 h, at 70 °C for 2 h and then at room temperature for 1 h. The static crystallization at 170 °C for 48 h was performed in a Teflon lined autoclave. The solid product was filtrated by centrifugation and washed with distilled water, and then dried at 60 °C for 12 h. The obtained composite was treated with HNO3 (2 mol L−1) at 60 °C for 2 h for two times to remove the Mg(OH)2 NCs.
The as-synthesized TS-1 and S-1 was dried at 120 °C for 12 h followed by calcinations at 550 °C for 8 h. S-1 was treated in aqueous solution of ammonium nitrate (7.5 wt%) for three times before catalytic reaction. The obtained TS-1 and S-1 catalyst samples was labeled with TS-1–0.05Mg/S-1–0.05Mg, TS-1–0.10Mg/S-1–0.10Mg, TS-1–0.30Mg/S-1–0.15Mg and TS-1–0.50Mg/S-1–0.25Mg, respectively.
2.2. Characterization
Powder X-ray diffraction (XRD) was performed on Panalytical X'Pert PRO diffractometer with Cu Kα1 (λ = 1.5406 Å) and a scanning rate of 1.2° min−1 in the 2 theta ranges from 5 to 80°. Transmission electron microscope (TEM) micrographs were obtained by JEOL JEM 2100 electron microscope operating at 200 kV. Fourier transform infrared (FT-IR) spectra were recorded on a Nicolet Spectrometer with a resolution of 4 cm−1. The self-supported wafers (about 10 mg cm−2) were placed in a cell with quartz windows, heated to 773 K and kept for 3 h under vacuum of 1 Pa. When the cell was cooled down to room temperature, the spectra were recorded. N2 sorption isotherms were measured on NOVA 1000e surface area and poresize analyzer (Quantachrome, USA) at 77 K. From the adsorption branch of isotherm curves at the P/P0 range between 0.001 and 0.10, the total specific surface areas (SBET) were calculated by the multi-point Brunauer–Emmett–Teller (BET) method. The micropore specific surface area (Smic) was calculated by Barrett–Joyner–Halenda (BJH) method. The external specific surface area (Sext) and external pore volume (Vext) were evaluated by the t-plot method. The total pore volume (Vtot) was determined from the amount adsorbed at the relative pressure of about 0.99.33 The chemical compositions of TS-1–xMg and S-1–xMg was measured by EDX in JSM-6700F.2.3. Catalytic performance
:nH2O2:nC4H10O:nC6H10O = 3:1.05:2.6:1. After 6 h, the product mixture was analyzed by a GC-7890 gas chromatograph equipped with a flame ionization detector (FID) and an OV-1701 capillary column.3. Results and discussion
3.1. Characterization of TS-1 and S-1
The Mg(OH)2 NCs possess lamellar structure with a size of 100–200 nm (seen in Fig. S1†).32 Due to abundant surface hydroxyl, the Mg(OH)2 NCs were dispersed into water to form a stable colloid. All precursors were mixed with the colloid to form a homogeneous synthesis mixture. Molecular sieve nanoslabs consequently generated and assembled around the Mg(OH)2 NCs in the aging process, resulting in a composite of molecular sieve and Mg(OH)2 NCs (Scheme 2). After the hydrothermal crystallization and removal of the entrapped Mg(OH)2 NCs to generate pores or caves, calcination was performed to provide TS-1 and S-1. For the obtained TS-1 and S-1 labeled with TS-1–0.05Mg/S-1–0.05Mg, TS-1–0.10Mg/S-1–0.10Mg, TS-1–0.30Mg/S-1–0.15Mg and TS-1–0.50Mg/S-1–0.25Mg, different amounts of predispersed Mg(OH)2 NCs were added into the synthesis mixture.TEM images (Fig. 1) show that TS-1 without Mg(OH)2 NCs exhibit a uniform rectangular plate shape with a comparatively smooth surface. With the increase of Mg(OH)2 NCs, the smooth surface of TS-1 become much more rough. When the additive amount of Mg(OH)2 NCs was 50 wt%, some large voids and caves are evidently located in the inner part of TS-1 crystals. The big voids and caves are useful to improve the diffusion properties and accessibility to active sites. S-1 without Mg(OH)2 NCs have twin crystal structures. In the presence of 10 wt% or 25 wt% Mg(OH)2 NCs, the twin crystal growth are obviously suppressed and the obtained S-1 also have much more rugged surface than those without Mg(OH)2 NCs. When the additive amount of Mg(OH)2 NCs was 25 wt%, a few of S-1 crystals have some obvious slits. These results reveal that Mg(OH)2 NCs have favorable interactions with silica species, affecting zeolite nucleation and growth via the attachment of Mg(OH)2 NCs onto the surface of TS-1 and S-1 crystals and entrapment inside TS-1 (seen in the Fig. S2†) and S-1 crystals.
 | ||
| Fig. 1 TEM images of TS-1–xMg and S-1–xMg: (a) TS-1, (b) TS-1–0.10Mg, (c) TS-1–0.50Mg, (d) S-1, (e) S-1–0.10Mg and (f) S-1–0.25Mg. | ||
The crystal phase structures of TS-1 and S-1 were characterized by XRD (Fig. 2). There are sharp peaks at 2θ = 7.9, 8.9, 23.0, 23.9 and 24.4° corresponding to the five characteristic peaks of MFI topology in all TS-1 and S-1. The TS-1–xMg crystals have an orthorhombic structure due to single peak at 2θ = 24.4°.34 While the diffraction double peaks at 2θ = 24.4° show that S-1–xMg possess a monoclinic structure after calcination.35,36 The relative intensity of peaks at 2θ = 7.9, 8.9, 23.0, 23.9 and 24.4° of TS-1–xMg/S-1–xMg both remarkably reduce when Mg(OH)2 NCs amount is 10 and 15 wt%, respectively. These results indicate that the Mg(OH)2 NCs influence the crystallization process of TS-1 and S-1 crystals. Moreover, XRD patterns of TS-1–xMg and S-1–xMg after removing Mg(OH)2 don't possess the characteristic peaks of Mg(OH)2 (at 2θ = 18.4 and 37.9°, seen in Fig. S3†).
 | ||
| Fig. 2 XRD patterns of (a) TS-1–xMg and (b) S-1–xMg synthesized in the presence of different Mg(OH)2 NCs amount. | ||
The textural properties of TS-1–xMg/S-1–xMg were measured by N2-sorption isotherms (Fig. 3). For TS-1–xMg/S-1–xMg, the nitrogen uptake at p/p0 ≈ 0.99 obviously both enhances with increasing Mg(OH)2 NCs additive amount. The hysteresis loops at p/p0 from 0.4 to 0.99 begins to appear for TS-1 synthesis in the presence of 30 wt% Mg(OH)2 NCs and become much more prominent when the Mg(OH)2 NCs is 50 wt%, showing the occurrence of mesopores in the TS-1–0.50Mg. No clear hysteresis loops are observed at high-pressure region for S-1–xMg.
 | ||
| Fig. 3 N2-sorption isotherms of (a) TS-1–xMg and (b) S-1–xMg synthesized in the presence of different Mg(OH)2 NCs amount. Each successive curve is offset by about 50 mL g−1 from its predecessor on the y-axis except N2-sorption isotherms of TS-1 and S-1. | ||
The textural parameters calculated from the nitrogen sorption isotherms are given in Table 1. For TS-1–xMg samples, with the increase of Mg(OH)2 NCs amount, external surface area gradually become larger and total pore volume increase. The total surface area first rise and then drop, up to the maximum for TS-1 synthesis in the presence of 10 wt% Mg(OH)2 NCs. The micropore surface area and volume (Vmic) change little when Mg(OH)2 amount increase from 0 to 10 wt%, and then drop with the further increase of Mg(OH)2. The total surface area and micropore surface area for S-1–xMg generally first rise and then drop, up to the maximum for S-1 synthesis in the presence of 5 wt% Mg(OH)2 NCs. The external surface area change little with the increase of Mg(OH)2 from 0 to 15%, but increase obviously when Mg(OH)2 amount is 25 wt%. These results indicate that the Mg(OH)2 NCs can affect the textural properties of TS-1 and S-1 and may play an important role as a hard template in the self-assembly process of MFI nanoslabs. The influence of Mg(OH)2 NCs on microstructure of S-1 is less than that of TS-1. The reason is that the S-1 crystal size synthesized by organic raw materials only has 200–300 nm, the Mg(OH)2 NCs with a size of 100–200 nm seems to be much larger as a hard template in the synthesis solution.
| Sample | SBET | Smic | Sext | Vtot | Vmic | Smic/SBET | Sext/SBET |
|---|---|---|---|---|---|---|---|
| (m2 g−1) | (cm3 g−1) | (%) | |||||
| TS-1 | 378 | 351 | 27 | 0.18 | 0.15 | 92.9 | 7.10 |
| TS-1–0.05Mg | 391 | 348 | 43 | 0.20 | 0.15 | 89.0 | 11.0 |
| TS-1–0.10Mg | 412 | 351 | 61 | 0.22 | 0.15 | 85.2 | 14.8 |
| TS-1–0.30Mg | 382 | 241 | 141 | 0.27 | 0.11 | 63.1 | 36.9 |
| TS-1–0.50Mg | 367 | 147 | 220 | 0.31 | 0.06 | 40.1 | 59.9 |
| S-1 | 362 | 296 | 66 | 0.19 | 0.11 | 81.8 | 18.2 |
| S-1–0.05Mg | 411 | 349 | 62 | 0.20 | 0.14 | 84.9 | 15.1 |
| S-1–0.10Mg | 361 | 317 | 45 | 0.18 | 0.12 | 87.8 | 12.2 |
| S-1–0.15Mg | 381 | 314 | 67 | 0.22 | 0.12 | 82.4 | 17.6 |
| S-1–0.25Mg | 345 | 218 | 127 | 0.22 | 0.09 | 63.2 | 36.8 |
The type and distribution of silanols in S-1 molecular sieves are of great significance for the catalytic performances in Beckmann rearrangement reaction.37 Treatment with nitrogen-containing basic solution often are performed to adjust the distribution of various silanols and construct catalytic active sites.38 As-synthesized S-1–xMg NCs were modified by NH4NO3 solution before being used as catalyst in the Beckmann rearrangement reaction. FT-IR spectra show that the NH4NO3 treatment constructed different silanols distribution of S-1–xMg (Fig. 4). Compared to the FT-IR spectrum of S-1 without NH4NO3 treatment, the 3740 cm−1 peak of terminal silanols is very weak for the S-1–xMg after NH4NO3 treatment. Two broad bands around 3680 and 3450 cm−1, corresponding to the vicinal silanols and silanol nests respectively, are much more prominent.
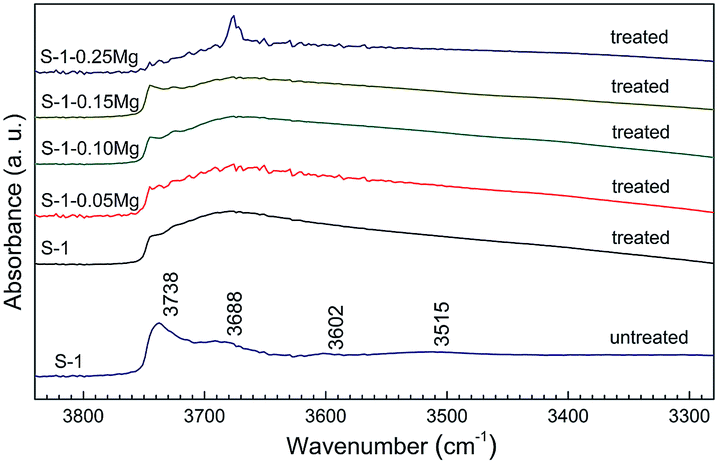 | ||
| Fig. 4 FT-IR spectra of S-1–xMg treated with NH4NO3. | ||
3.2. Catalytic performance of TS-1 and S-1 catalysts
 | ||
| Fig. 5 The catalytic performance of TS-1–xMg in batch cyclohexanone ammoximation. | ||
 | ||
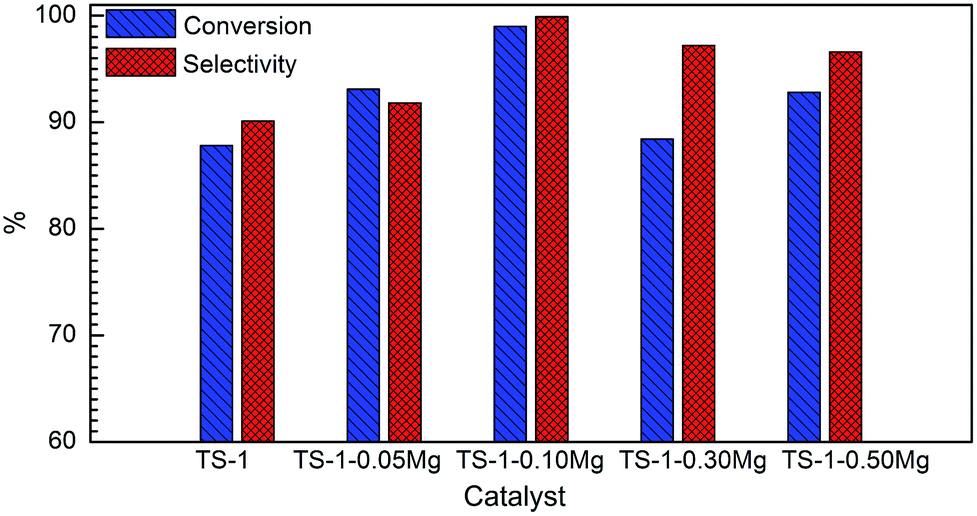
| Fig. 6 The conversion of cyclohexanone oxime (a) and selectivity of ε-caprolactam (b) with S-1–xMg as catalysts in the vapor phase Beckmann rearrangement of cyclohexanone oxime with WHSV = 3 h−1. | ||
It has been suggested the vicinal silanols and silanol nests are favourable species for the formation of ε-caprolactam in the vapor phase Beckmann rearrangement, while the terminal silanols are suitable for the formation of by-products.37,43,44 After NH4NO3 treatment, most of the terminal silanols were removed. The vicinal silanols and silanol nests on all the catalysts appeared in large quantity (Fig. 4). This change should be the reason why the S-1–xMg samples gave nearly complete conversion of cyclohexanone oxime and high selectivity to ε-caprolactam. For all S-1–xMg catalysts, the abundant vicinal silanols and silanol nests were both obtained after NH4NO3 treatment. The different catalytic performances of S-1–xMg catalysts should be attributed to their different textural properties. The larger ratio external surface area facilitate the mass diffusion of reactants and products, while abundant vicinal silanols and silanol nests located intensively in the inner part of S-1 are the active sites of Beckmann rearrangement. Too much external surface may reduce the active sites in the inner pores. Thus, moderate external and internal surface area are the best propitious to the reaction, which are the reason why catalytic performance for the S-1 synthesized with Mg(OH)2 NCs except S-1–0.25Mg catalyst was higher than those without Mg(OH)2 NCs. The lower catalytic performance of S-1–0.25Mg catalyst should be due to the excess Mg(OH)2 NCs which have not been removed completely by HNO3 and its lower relative crystalline degree compared to those of S-1 catalysts prepared with smaller amount of Mg(OH)2 NCs. The excess Mg(OH)2 NCs influenced the reaction environment. The crystallization of S-1 was retarded to some extent. The low relative crystalline degree means the reduction of active sites in S-1 catalyst.
Interestingly, the corresponding catalytic performance has been remarkable improved and the maximum activity is obtained when external surface area proportion in the total specific surface area is in the range from 12% to 15%, regardless of TS-1 or S-1. It can be understood that the utility of moderate amount Mg(OH)2 NCs greatly reduce diffusion resistance and properly improve the utilization ratio of active sites for cyclohexanone ammoximation and cyclohexanone oxime Beckmann rearrangement. But due to the formation of large voids and cracks, too many NCs make significant reduction in the total number of active sites, which were mainly located in the micropores.
4. Conclusions
In conclusion, introducing Mg(OH)2 NCs into the synthesis solution can effectively control the microstructure of TS-1 and S-1 molecular sieve crystals and remarkably improve their catalytic performances. The obtained TS-1 and S-1 crystals possessed much more rugged surface, some voids and slits existed inside the crystals. With increasing Mg(OH)2 NCs addition amount, the total surface area first rise and then drop, external surface area of TS-1–xMg gradually increased. These are in favor of the mass transfer and accessible to active sites, thus improve the catalytic performance of cyclohexanone ammoximation reaction. After different amounts of Mg(OH)2 NCs were added into the S-1 synthesis solution, the total surface area and micropore surface area generally first rise and then drop. The catalytic performances of S-1 in the vapor phase Beckmann rearrangement also were improved. For TS-1–xMg and S-1–xMg, the best catalysts were both obtained with 10 wt% Mg(OH)2 NCs, the micropore surface area and external surface area were in accord with the best match for the reaction, reducing diffusion resistance while improving the utilization of the active sites. The introduction of inorganic NCs as template will find its application in the microstructure- and morphology-controllable synthesis of other molecular sieves for superior catalytic performances.Acknowledgements
Financial supports from the National Natural Science Foundation of China (no. U1204203, 21401168), Innovation Fund for Elitists of Henan Province, China (no. 0221001200) and China Postdoctoral Science Foundation (no. 2013T60705) are acknowledged.Notes and references
- D. P. Serrano, J. M. Escola and P. Pizarro, Chem. Soc. Rev., 2013, 42, 4004–4035 RSC.
- M. Choi, K. Na, J. Kim, Y. Sakamoto, O. Terasaki and R. Ryoo, Nature, 2009, 461, 246–249 CrossRef CAS PubMed.
- C. Perego and R. Millini, Chem. Soc. Rev., 2013, 42, 3956–3976 RSC.
- L. Chen, Y. M. Wang and M. Y. He, Mater. Res. Bull., 2011, 46, 698–701 CrossRef CAS PubMed.
- H. Li, J. J. Jin, W. Wu, C. C. Chen, L. Li, Y. S. Li, W. R. Zhao, J. L. Gu, G. R. Chen and J. L. Shi, J. Mater. Chem., 2011, 21, 19395–19401 RSC.
- J. C. Lin and M. Z. Yates, Langmuir, 2005, 21, 2117–2120 CrossRef CAS PubMed.
- S. Lee and D. F. Shantz, Chem. Commun., 2004, 680–681 RSC.
- B. Y. Liu, Q. Q. Duan, C. Li, Z. H. Zhu, H. X. Xi and Y. Qian, New J. Chem., 2014, 38, 4380–4387 RSC.
- D. Nandan, S. K. Saxena and N. Viswanadham, J. Mater. Chem. A, 2014, 2, 1054–1059 CAS.
- H. L. Chen, J. Ding and Y. M. Wang, New J. Chem., 2014, 38, 308–316 RSC.
- Y. R. Wang, M. Lin and A. Tuel, Microporous Mesoporous Mater., 2007, 102, 80–85 CrossRef CAS PubMed.
- C. S. Mei, Z. C. Liu, P. Y. Wen, Z. K. Xie, W. M. Hua and Z. Gao, J. Mater. Chem., 2008, 18, 3496–3500 RSC.
- Y. M. Fang, H. Q. Hu and G. H. Chen, Chem. Mater., 2008, 20, 1670–1672 CrossRef CAS.
- J. H. Jacobsen, C. Madsen, J. Houzvicak, I. Schmidt and A. Carlsson, J. Am. Chem. Soc., 2000, 122, 7116–7117 CrossRef.
- Y. S. Tao, H. Kanoh and K. Kaneko, J. Am. Chem. Soc., 2003, 125, 6044–6045 CrossRef CAS PubMed.
- W. C. Li, A. H. Lu, R. Palkovits, W. Schmidt, B. Spliethoff and F. Schüth, J. Am. Chem. Soc., 2005, 127, 12595–12600 CrossRef CAS PubMed.
- F. N. Gu, F. Wei, J. Y. Yang, N. Lin, W. G. Lin, Y. Wang and J. H. Zhu, Chem. Mater., 2010, 22, 2442–2450 CrossRef CAS.
- B. Li, Z. J. Hu, B. Kong, J. X. Wang, W. Li, Z. K. Sun, X. F. Qian, Y. S. Yang, W. Shen, H. L. Xu and D. Y. Zhao, Chem. Sci., 2014, 5, 1565–1573 RSC.
- K. Möller, B. Yilmaz, R. M. Jacubinas, U. Müller and T. Bein, J. Am. Chem. Soc., 2011, 133, 5284–5295 CrossRef PubMed.
- L. Lakiss, M. Rivallan, J. M. Goupil, J. El Fallah and S. Mintova, Catal. Today, 2011, 168, 112–117 CrossRef CAS PubMed.
- C. Burda, X. Chen, R. Narayanan and M. A. El-Sayed, Chem. Rev., 2005, 105, 1025–1102 CrossRef CAS PubMed.
- H. B. Zhu, Z. C. Liu, Y. D. Wang, D. J. Kong, X. H. Yuan and Z. K. Xie, Chem. Mater., 2008, 20, 1134–1139 CrossRef CAS.
- A. Corma and H. Garcia, Chem. Rev., 2003, 103, 4307–4366 CrossRef CAS PubMed.
- G. Dahlfoff, J. P. M. Niederer and W. F. Hölderich, Catal. Rev., 2001, 43, 381–441 Search PubMed.
- M. Kitammura, H. Ichihashi and H. Tojima, US 5403801, 1995.
- R. Mokaya and M. Poliakoff, Nature, 2005, 437, 1243–1244 CrossRef CAS PubMed.
- Y. Izumi, H. Ichihashi, Y. Shimazu, M. Kitamura and H. Sato, Bull. Chem. Soc. Jpn., 2007, 80, 1280–1287 CrossRef CAS.
- Y. Xue, Y. L. Xie, H. J. Wei, Y. Q. Wen, X. Y. Wang and B. j. Li, New J. Chem., 2014, 38, 4229–4234 RSC.
- L. Xu, J. H. Ding, Y. L. Yang and P. Wu, J. Catal., 2014, 309, 1–10 CrossRef CAS PubMed.
- Y. Q. Deng, S. F. Yin and C. T. Au, Ind. Eng. Chem. Res., 2012, 51, 9492–9499 CrossRef CAS.
- J. Kim, W. Park and R. Ryoo, ACS Catal., 2011, 1, 337–341 CrossRef CAS.
- B. J. Li, H. Q. Cao and G. Yin, J. Mater. Chem., 2011, 21, 13765–13768 RSC.
- M. Jaroniec, M. Kruk and J. P. Olivier, Langmuir, 1999, 15, 5410–5413 CrossRef CAS.
- X. X. Wu, Y. Q. Wang, T. Zhang, S. H. Wang, P. X. Yao, W. P. Feng, Y. Lin and J. Xu, Catal. Commun., 2014, 50, 59–62 CrossRef CAS PubMed.
- A. Zecchina, S. Bordiga, G. Spoto and L. Marchese, J. Phys. Chem., 1992, 96, 4985–4990 CrossRef CAS.
- F. H. Meng, Y. Q. Wang, L. N. Wang, R. M. Yang and T. Zhang, J. Mol. Catal. A: Chem., 2011, 335, 105–111 CrossRef CAS PubMed.
- G. P. Heitmann, G. Dahlfoff and W. F. Hölderich, J. Catal., 1999, 186, 12–19 CrossRef CAS.
- H. Ichihashi, K. Sugita and M. Yako, US 6303099, 2001.
- J. Kärger and R. Valiullin, Chem. Soc. Rev., 2013, 42, 4172–4197 RSC.
- H. Xu, Y. T. Zhang, H. H. Wu, Y. M. Liu, X. H. Li, J. G. Jiang, M. Y. He and P. Wu, J. Catal., 2011, 281, 263–272 CrossRef CAS PubMed.
- S. T. Tsai, P. Y. Chao, T. C. Tsai, I. Wang, X. X. Liu and X. W. Guo, Catal. Today, 2009, 148, 174–178 CrossRef CAS PubMed.
- Y. Zuo, W. C. Song, C. Y. Dai, Y. P. He, M. L. Wang, X. S. Wang and X. W. Guo, Appl. Catal., A, 2013, 453, 272–279 CrossRef CAS PubMed.
- G. P. Heitmann, G. Dahlhoff and W. F. Hölderich, Appl. Catal., A, 1999, 185, 99–108 CrossRef CAS.
- G. P. Heitmann, G. Dahlhoff, J. P. M. Niederer and W. F. Hölderich, J. Catal., 2000, 194, 122–129 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra08277j |
| This journal is © The Royal Society of Chemistry 2015 |