
Molecular asphaltene models based on Clar sextet theory†
Abstract
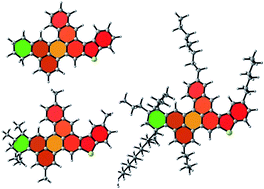
Asphaltenes, the base components in asphalt used for pavements and roads, are high-molecular-weight polycyclic aromatic hydrocarbons with long aliphatic chains and polar functionalities. Significant efforts have been made to develop representative asphalt models for molecular simulations, and several molecular structures have been proposed for asphaltenes based on experimental results and theoretical predictions. Many properties, such as viscosity, depend on the underlying detailed chemical structure of asphaltenes, since it defines the interactions at the nanoscale that dominate the formation of nanoaggregates. Among these interactions, π–π stacking plays a central role in the formation of clusters at the nanoscale level, with strong dependence on the electronic arrangement of π electrons in the polycyclic aromatic core of asphaltenes. Here, based on Clar sextet theory and density functional theory (DFT) calculations, we propose asphaltene molecules that optimize the distribution of π electrons in the polycyclic aromatic core and minimize geometrical strain within the molecule. Accordingly, more stable isomers than the asphaltene molecules used in current asphalt models are proposed. These new isomers include a more representative arrangement of the π electrons, crucial for further description of intermolecular interactions and formation of nanoaggregates. Considering these basic guidelines in future molecular modeling of asphalt will improve the development of more accurate models that describe the behavior of these complex materials at the nanoscale.
 Please wait while we load your content...
Please wait while we load your content...

