
UV-patternable nanocomposite containing CdSe and PbS quantum dots as miniaturized luminescent chemo-sensors†
Abstract
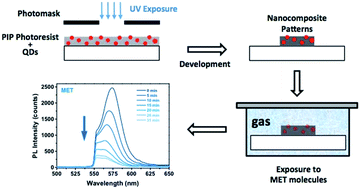
In this study, a novel multifunctional hybrid polymer-based luminescent material, particularly formulated for photolithography, was developed, fabricated and tested as a miniaturized chemosensor. This nanocomposites were formulated with either luminescent CdSe (for the visible) or PbS (for the near-IR) colloidal QDs embedded in a polyisoprene-based photoresist (PIP). We checked the sensing capability of the nanocomposite by exposing 1 cm2 CdSe nanocomposite patterns to vapours of some analyte solutions such as 2-mercaptoethanol (MET) and ethylenediamine (EDA). The transduction mechanism of the sensor is based on changes of the QD photoluminescence (PL) when molecules are adsorbed on the QD surface. Because the polymer used suffered from swelling during the developmental step of the sensor fabrication, the diffusion of the analytes through the matrix was rather high. As a result, the sensor response to the analyte–QD interactions was considerably short and sensitive. We observed shorter sensor response times for MET than EDA. Moreover, we found a limit of detection of MET and EDA of 0.1 pg and 15 ng, respectively. The linear detection range for MET and EDA was determined to be over an analyte concentration of 6 and 5 orders of magnitude, respectively. We also tested the PbS-based nanocomposite response to MET and EDA and found very different responses. Although EDA quenched the PbS PL, exposure to MET molecules resulted in a 6.5-fold enhancement of the PL. The mechanisms of the observed effects are discussed in detail.
 Please wait while we load your content...
Please wait while we load your content...

