DOI:
10.1039/C5QO00250H
(Research Article)
Org. Chem. Front., 2015,
2, 1485-1499
Stereoselective formation of chiral trans-4-hydroxy-5-substituted 2-pyrrolidinones: syntheses of streptopyrrolidine and 3-epi-epohelmin A†
Received
8th August 2015
, Accepted 3rd September 2015
First published on 7th September 2015
Abstract
A highly diastereoselective approach for the synthesis of trans-4-hydroxy-5-substituted 2-pyrrolidinones has been developed through an intramolecular cascade process of α-chiral aldimines using alkyl, aryl, alkynyl, and alkenyl Grignard reagents. The stereochemistry at the C-5 position of 2-pyrrolidinone after reaction with alkyl, aryl, and alkenyl Grignard reagents was solely controlled by α-alkoxy substitution. For alkynyl Grignard reagents, the stereochemistry was controlled by coordination of the α-alkoxy substitution and the stereochemistry of the sulfinamide. The utility of this one-pot cascade protocol is demonstrated by the asymmetric synthesis of streptopyrrolidine 5 and 3-epi-epohelmin A 3-epi-6.
Introduction
The development of efficient and stereoselective reactions and methodologies for carbon–carbon bond formation is one of the major pursuits in organic synthesis and medicinal chemistry.1 Ellman and Davis reported, respectively, an effective method to prepare chiral amines through the addition reaction of imines bearing chiral auxiliaries (e.g., N-tert-butanesulfinamide and N-toluenesulfinamide) with nucleophilic reagents.2,3 Nowadays, the addition of corresponding Grignard reagents to chiral N-tert-butanesulfinyl imines is widely applied in organic synthesis.4 Our group has also explored the utility of chiral N-tert-butanesulfinyl imines, and successfully accomplished the divergent synthesis of several bioactive natural products.5 In addition, we observed an interesting stereoselective tert-butyl migration from sulfur to carbon when N-tert-butanesulfinyl iminoacetate was treated with various benzylzinc bromides.6
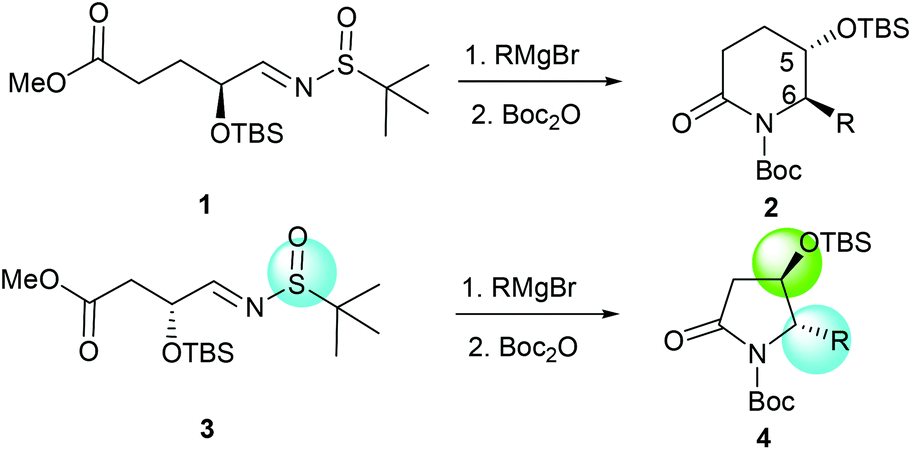
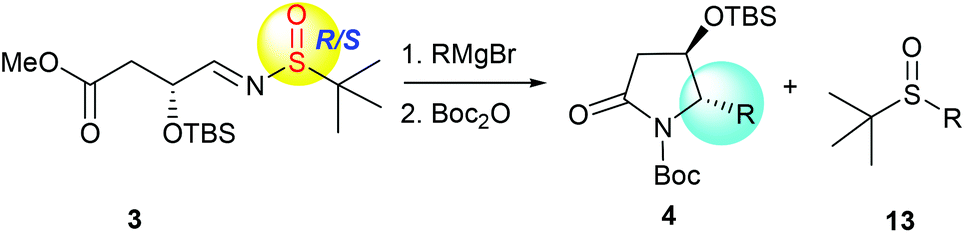
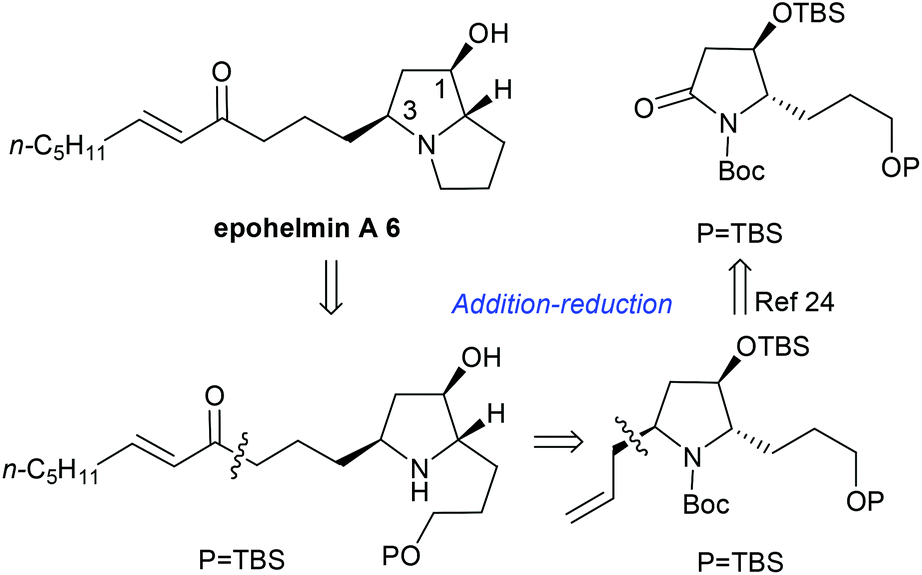
More recently, we discovered an intramolecular cascade process for the highly diastereoselective synthesis of versatile trans-5-hydroxy-6-substituted 2-piperidinones 2.7 In this case, the stereoselectivity of C-5 and C-6 strongly favored the trans form and the new stereogenic center at the C-6 position was solely controlled by the tert-butyldimethylsilyl ether (α-OTBS) group of imine 1. Encouraged by this highly stereoselective imine addition and the subsequent in situ formation of 2-piperidinones 2, as a continuous work, we became interested in a similar imine substrate 3 with one less carbon, which would potentially produce trans-4-hydroxy-5-substituted 2-pyrrolidinones 4 (Fig. 1).
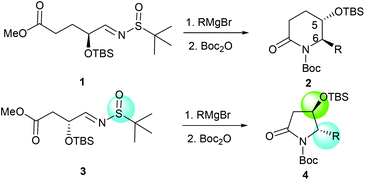 |
| | Fig. 1 Formation of chiral 2-piperidinones and 2-pyrrolidinones. | |
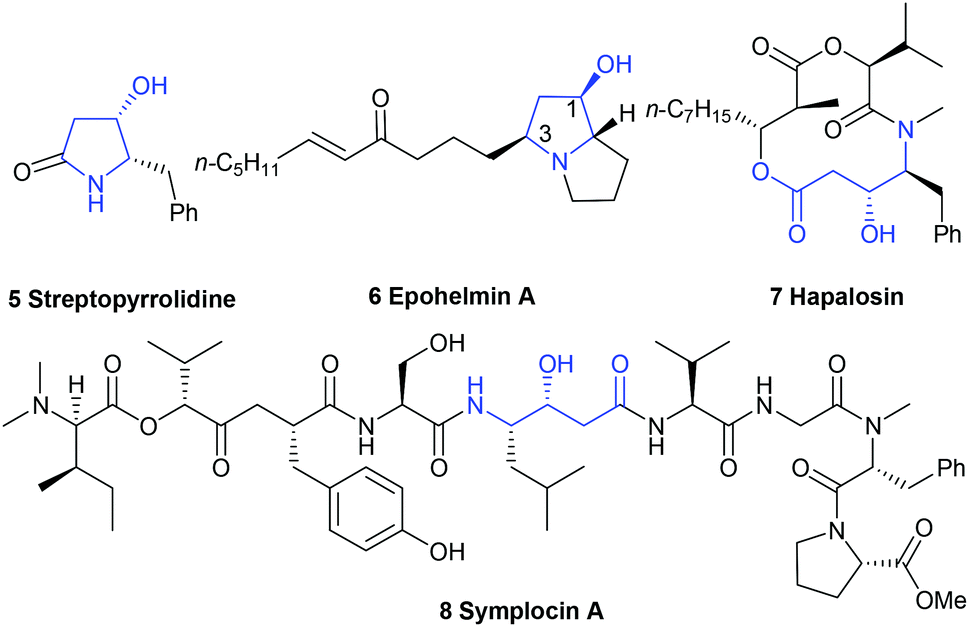
Chiral functionalized trans-4-hydroxy-5-substituted 2-pyrrolidinones and the ring-opened form γ-amino-β-hydroxybutyric acids are present as a core structure or subfeature in many biologically relevant alkaloids8,9 isolated from marine and terrestrial plants and animals, as well as in pharmaceutical agents.10 For example, streptopyrrolidine 5, isolated from the fermentation broth of marine Streptomyces sp. KORDI-3973,11 could significantly block the capillary tube formation of cells at the same potency as the known angiogenesis inhibitor SU11248, whereas epohelmin A 6, isolated from an unidentified fungus (strain FKI-0929), could inhibit recombinant lanosterol synthase (IC50 = 10 μM).12 Ring-opened form, (3R,4S)-γ-amino-β-hydroxybutyric acid unit,13 exists in hapalosin 7 and symplocin A 8. The former showed multi-drug resistance reversing activity in cancer cells,14 and the latter displayed very potent cathepsin E inhibition (IC50 = 300 pM)15 (Fig. 2).
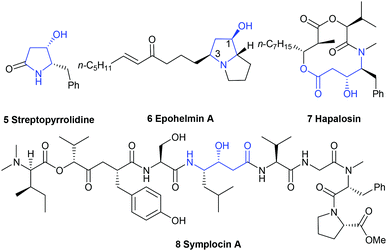 |
| | Fig. 2 The structure of several bioactive molecules. | |
In the past few decades, a wide range of synthetic methods for the stereoselective construction of trans-4-hydroxyl-5-substituted 2-pyrrolidinones 4 and the ring-opened form have been developed.9 The common approaches among them are based on reductive alkylation9d–f,k and nucleophilic substitution,9c which usually require multiple steps and lead to an unsatisfactory overall yield. Therefore, an efficient and stereoselective approach for direct preparation of 4 remains a challenge.16 Herein we report our results of the one-pot cascade process starting from α-chiral aldimine 3 and Grignard reagents to generate chiral 2-pyrrolidinones 4, as well as the application in the asymmetric syntheses of streptopyrrolidine 5 and 3-epi-epohelmin A (3-epi-6).
Results and discussion
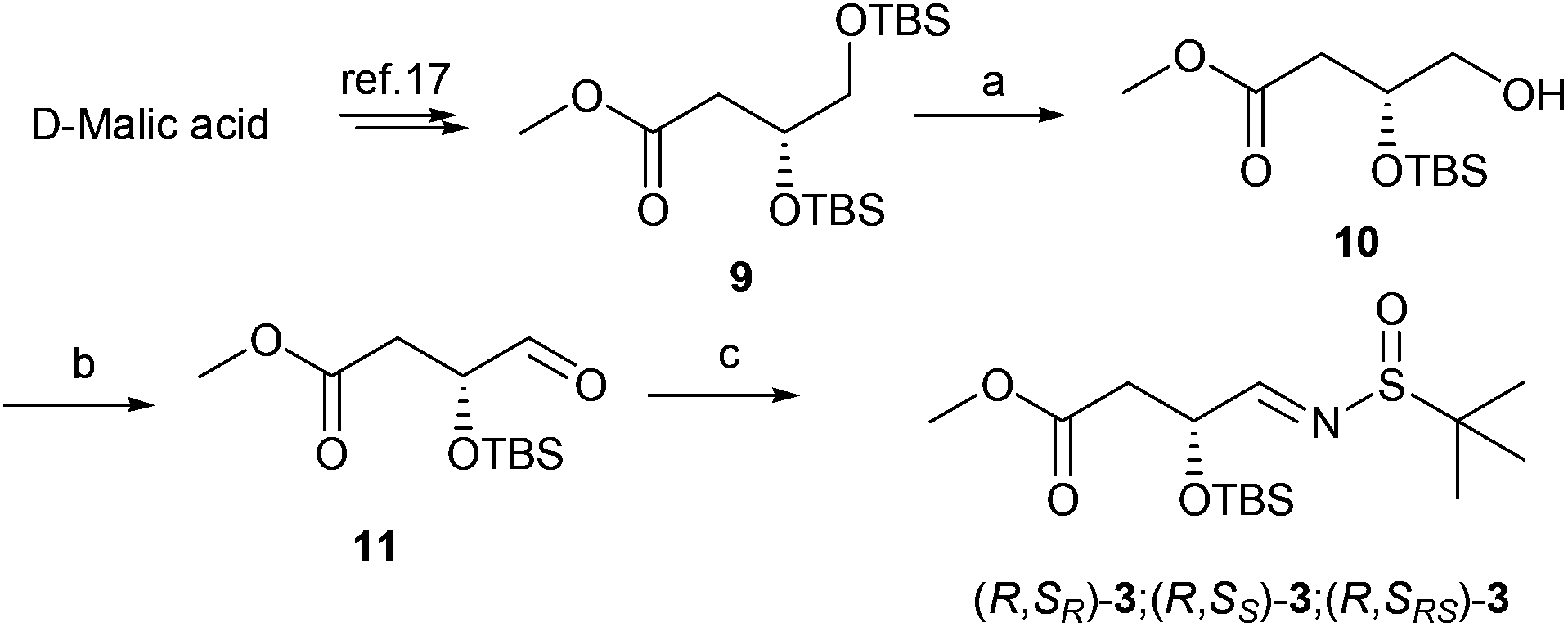
The synthesis of aldimine 3 is demonstrated in Scheme 1. D-Malic acid was conveniently converted to ester 9 according to the known procedure in 74% overall yield.17 The subsequent regioselective desilylation from the primary hydroxyl group was achieved by controlling the reaction temperature at −40 °C in the presence of camphorsulfonic acid (CSA) to produce alcohol 10 in 65% yield. Oxidation of 10 with Dess–Martin periodinane (DMP)18 and subsequent condensation with 2-methylpropane-2-sulfinamide in the presence of anhydrous copper sulfate19 led to N-tert-butanesulfinyl imines 3 in 84% overall yield (Scheme 1).
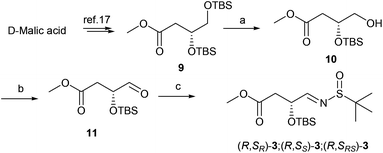 |
| | Scheme 1 Preparation of N-tert-butanesulfinyl aldimines 3. Reagents and conditions: (a) CSA, MeOH/DCM, −40 °C, 8 h, 65%; (b) DMP, DCM, room temperature (rt), 0.5 h, quantitative yield; (c) 2-methylpropane-2-sulfinamide, CuSO4, pyridinium p-toluenesulfonate, DCM, 24 h, 84%. | |
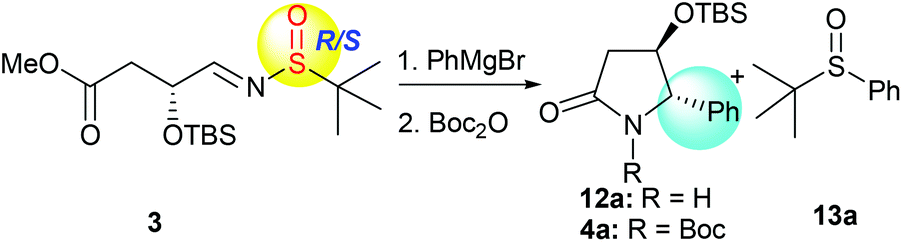
With the desired precursor 3 in hand, we started to investigate the intramolecular tandem sequence by reacting it with Grignard reagents. When optically pure (R,SR)-3 was treated with phenyl magnesium bromide at −78 °C, the desired product 12a was obtained. Because of its co-elution with the sulfoxide by-product 13a in silica gel chromatography, the lactam-NH was protected as the N-tert-butoxycarbonyl (N-Boc) form, and the pure imide 4a was obtained with high diastereoselectivity (dr = 99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) in spite of low yield (40%, Table 1, entry 1). Like the tandem process involving imine 1,7 the yield of 4a was greatly improved to 74% when the reaction was conducted from −78 °C to room temperature and maintained at room temperature overnight, the diastereoselectivity remained unchanged (dr = 99:1, Table 1, entry 2). Regardless of the chirality of sulfur in the imine substrate, both (R,SS)-3 and (R,SR)-3, as well as (R,SRS)-3, led to the same product 4a with high diastereoselectivities (dr = 99:1, Table 1, entries 2–4), indicating that the stereochemistry of the new chiral center at C-5 was solely controlled by the α-OTBS group. Use of different solvents was also investigated for the reaction with (R,SRS)-3, and the results are summarized in Table 1 (entries 5–7). The reaction in tetrahydrofuran (THF) offered better conversion than in tetrahydrofuran/dichloromethane (Table 1, entries 4 and 5). Different amounts of Grignard reagents were also assessed. The use of less than three equivalents of phenylmagnesium bromide also produced the desired product 4a with high diastereoselectivity, albeit in much lower yields (Table 1, entries 8 and 9).
:1) in spite of low yield (40%, Table 1, entry 1). Like the tandem process involving imine 1,7 the yield of 4a was greatly improved to 74% when the reaction was conducted from −78 °C to room temperature and maintained at room temperature overnight, the diastereoselectivity remained unchanged (dr = 99:1, Table 1, entry 2). Regardless of the chirality of sulfur in the imine substrate, both (R,SS)-3 and (R,SR)-3, as well as (R,SRS)-3, led to the same product 4a with high diastereoselectivities (dr = 99:1, Table 1, entries 2–4), indicating that the stereochemistry of the new chiral center at C-5 was solely controlled by the α-OTBS group. Use of different solvents was also investigated for the reaction with (R,SRS)-3, and the results are summarized in Table 1 (entries 5–7). The reaction in tetrahydrofuran (THF) offered better conversion than in tetrahydrofuran/dichloromethane (Table 1, entries 4 and 5). Different amounts of Grignard reagents were also assessed. The use of less than three equivalents of phenylmagnesium bromide also produced the desired product 4a with high diastereoselectivity, albeit in much lower yields (Table 1, entries 8 and 9).
Table 1 Optimization of the tandem process
|
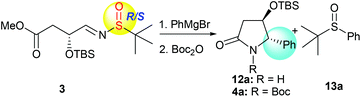
|
| Entrya |
Imine |
T (°C) |
Solvent |
Yieldb (%) |
trans:cisc |
|
The reactions were performed with α-chiral substituted aldimines 3 (1.0 mmol), phenyl magnesium bromide (3 mL, 1.0 M in THF) from −78 °C to rt overnight, crude product was treated with Boc2O (2.0 mmol), DMAP (1.0 mmol), and triethylamine (5.0 mmol) in DMF for 24 h.
Isolated yield.
Determined by HPLC or 1H NMR.
Phenyl magnesium bromide (1 mL, 1.0 M in THF).
Phenyl magnesium bromide (2 mL, 1.0 M in THF).
|
| 1 |
(R,SR)-3 |
−78 |
THF |
40 |
99:1 |
| 2 |
(R,SR)-3 |
−78 to rt |
THF |
74 |
99:1 |
| 3 |
(R,SS)-3 |
−78 to rt |
THF |
69 |
99:1 |
| 4 |
(R,SRS)-3 |
−78 to rt |
THF |
71 |
99:1 |
| 5 |
(R,SRS)-3 |
−78 to rt |
THF/DCM (3/5) |
45 |
99:1 |
| 6 |
(R,SRS)-3 |
−78 to rt |
THF/Et2O (3/5) |
62 |
99:1 |
| 7 |
(R,SRS)-3 |
−78 to rt |
THF/PhMe (3/5) |
65 |
99:1 |
| 8d |
(R,SRS)-3 |
−78 to rt |
THF |
25 |
99:1 |
| 9e |
(R,SRS)-3 |
−78 to rt |
THF |
47 |
99:1 |
Next, we turned our attention to investigate the scope and limitation of this method in the preparation of trans-4-hydroxy-5-substituted 2-pyrrolidinones. A survey of different Grignard reagents was conducted by reaction with (R,SRS)-3 under the established optimal conditions, as summarized in Table 2. When substituted phenyl Grignard reagents were used, the intramolecular tandem addition–cyclization proceeded smoothly with high diastereoselectivities and in excellent yields (Table 2, entries 2–9) except that the m-fluoro-phenylmagnesium reagent led to a slightly lower diastereoselectivity of the desired product 4f (Table 2, entry 6). Bicyclic Grignard reagents, including α- and β-naphthyl magnesium bromides, also afforded the desired lactams 4j,k, and the less hindered β-naphthyl magnesium bromide provided better diastereoselectivity and higher yield for this tandem process (Table 2, entries 10 and 11). Several alkyl-chain Grignard reagents were also investigated, and the yields of products 4l–p were slightly lower than those of substituted phenyl Grignard reagents (Table 2, entries 12–16). It is noteworthy that saturated cyclic Grignard reagents also gave the desired lactams 4q,r in 66–68% yield (Table 2, entries 17 and 18). Although benzylmagnesium bromide could give the corresponding product 4s in 70% yield (Table 2, entry 19), the reaction with allylMgBr was very messy (Table 2, entry 20). All the above-mentioned results indicate that the tandem sequence of α-chiral aldimine 3 was quite suitable for different substituted alkyl and aryl Grignard reagents and the stereogenic center at C-5 was solely controlled by the α-OTBS group.
Table 2 Reactions of different Grignard reagents with (R,SRS)-3
The stereochemistry of product 4a was unambiguously determined as the trans form by X-ray crystallography.26 Other compounds 4b–s were accordingly assigned as the trans forms based on the similar coupling constant (J) between the protons C4-H (C![[H with combining low line]](https://www.rsc.org/images/entities/i_char_0048_0332.gif) -OTBS) and C5-H (C-alkyl or aryl).
-OTBS) and C5-H (C-alkyl or aryl).
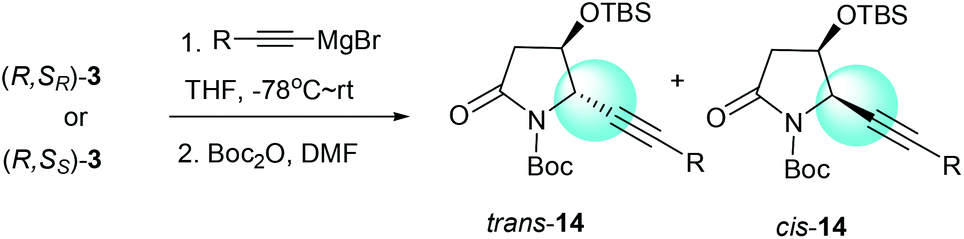
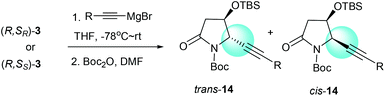
We had previously demonstrated that the reactions of α-chiral aldimines 1 with alkynyl Grignard reagents could lead to 2-piperidinones 2 with different stereoselectivities depending on the chirality of sulfur.7b The α-chiral aldimine 3, with one less carbon in the chain, also performed in a similar way. When (R,SR)-3 was treated with (2-phenylethynyl)magnesium bromide and followed by N-Boc protection, the desired imide 14a was produced in 76% yield with high diastereoselectivity (dr = 99:1) (Table 3, entry 1). Other aryl- and alkyl-substituted alkynyl Grignard reagents were also investigated for the reaction with (R,SR)-3, and the desired imides 14b–e were produced with high diastereoselectivities (dr = 99:1) (Table 3, entries 2–5). The reactions of α-chiral aldimine (R,SS)-3 with alkynyl Grignard reagents also afforded the desired products in comparable combined yields of both trans and cis isomers, but with significantly lower diastereoselectivities. When (R,SS)-3 was treated with (2-phenylethynyl)magnesium bromide, the desired imide 14a was obtained in 73% combined yield with low diastereoselectivity (dr = 38:62), favoring the cis form (Table 3, entry 6). Both (2-m-tolylethynyl)magnesium bromide and [2-(4-fluorophenyl)ethynyl]magnesium bromide also yielded the imides 14b and 14c with low diastereoselectivities, predominantly in the cis forms (Table 3, entries 7 and 8). Interestingly, when (3,3-dimethylbut-1-ynyl)magnesium bromide and hept-1-ynylmagnesium bromide were subjected to the reaction sequence, the major isomer of the resulting imides 14d and 14e were the trans forms (Table 3, entries 9 and 10). Our results indicate that the stereochemistry outcome of the reactions with alkynyl Grignard reagents was controlled by both the α-alkoxy substitution and the chiral sulfinamide. When α-chiral aldimine (R,SR)-3 was used, imide 14 was obtained with high diastereoselectivity (dr = 99:1) in the trans form. However, the reactions with α-chiral aldimine (R,SS)-3 gave poor diastereoselectivities, with the aryl-substituted alkynyl Grignard reagents producing the major imides 14 in the cis forms and alkyl-substituted alkynyl Grignard reagents favoring trans products.
Table 3 Reactions of different alkynyl Grignard reagents with 3
|
|
| Entrya |
R |
3
|
14a–e
|
Yieldb (%) |
trans:cisc |
|
Reactions were performed with 3 (1.0 mmol), Grignard reagents (3 mL, 1.0 M in THF) in dry THF (5 mL) at −78 °C to rt overnight, crude product was treated with Boc2O (2.0 mmol), DMAP (1.0 mmol), and triethylamine (5.0 mmol) in DMF for 24 h.
Isolated yield.
Determined by 1H NMR.
|
| 1 |
C6H5 |
(R,SR) |
(2S,3R)-14a |
76 |
99:1 |
| 2 |
3-Me-C6H4 |
(R,SR) |
(2S,3R)-14b |
80 |
99:1 |
| 3 |
4-F-C6H4 |
(R,SR) |
(2S,3R)-14c |
77 |
99:1 |
| 4 |
CH3(CH2)3CH2 |
(R,SR) |
(2S,3R)-14d |
79 |
99:1 |
| 5 |
C(CH3)3 |
(R,SR) |
(2S,3R)-14e |
70 |
99:1 |
| 6 |
C6H5 |
(R,SS) |
(2R,3R)-14a |
73 |
38:62 |
| 7 |
3-Me-C6H4 |
(R,SS) |
(2R,3R)-14b |
75 |
35:65 |
| 8 |
4-F-C6H4 |
(R,SS) |
(2R,3R)-14c |
72 |
34:66 |
| 9 |
CH3(CH2)3CH2 |
(R,SS) |
(2R,3R)-14d |
71 |
61:39 |
| 10 |
C(CH3)3 |
(R,SS) |
(2R,3R)-14e |
63 |
68:32 |
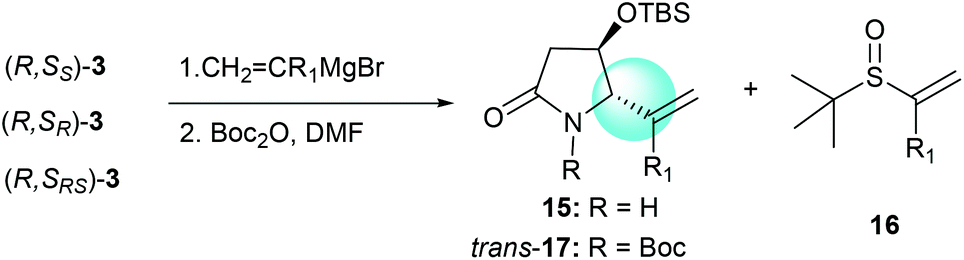
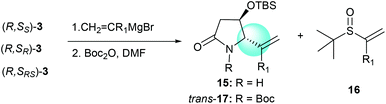
The tandem process of reacting α-chiral aldimine 3 with alkenyl Grignard reagents was also investigated. Like the reaction with imine substrate 1,7b the generation of the stereogenic center at C-5 was solely controlled by the α-OTBS group (Table 4, entries 1–5).
Table 4 Reactions of different alkenyl Grignard reagents with 3
|
|
| Entrya |
3
|
R1 |
17
|
Yieldb (%) |
trans:cisc |
|
Reactions were performed with α-chiral substituted aldimines 3 (1.0 mmol), alkenyl Grignard reagents (3 mL, 1.0 M in THF) in dry THF (5 mL) at −78 °C to rt overnight, crude product was treated with Boc2O (2.0 mmol), DMAP (1.0 mmol), and triethylamine (5.0 mmol) in DMF for 24 h.
Isolated yield.
Determined by HPLC or 1H NMR.
|
| 1 |
(R,SRS) |
H |
17a
|
69 |
99:1 |
| 2 |
(R,SR) |
H |
17a
|
74 |
99:1 |
| 3 |
(R,SS) |
H |
17a
|
65 |
99:1 |
| 4 |
(R,SR) |
Et- |
17b
|
68 |
98:2 |
| 5 |
(R,SS) |
Et- |
17b
|
59 |
96:4 |
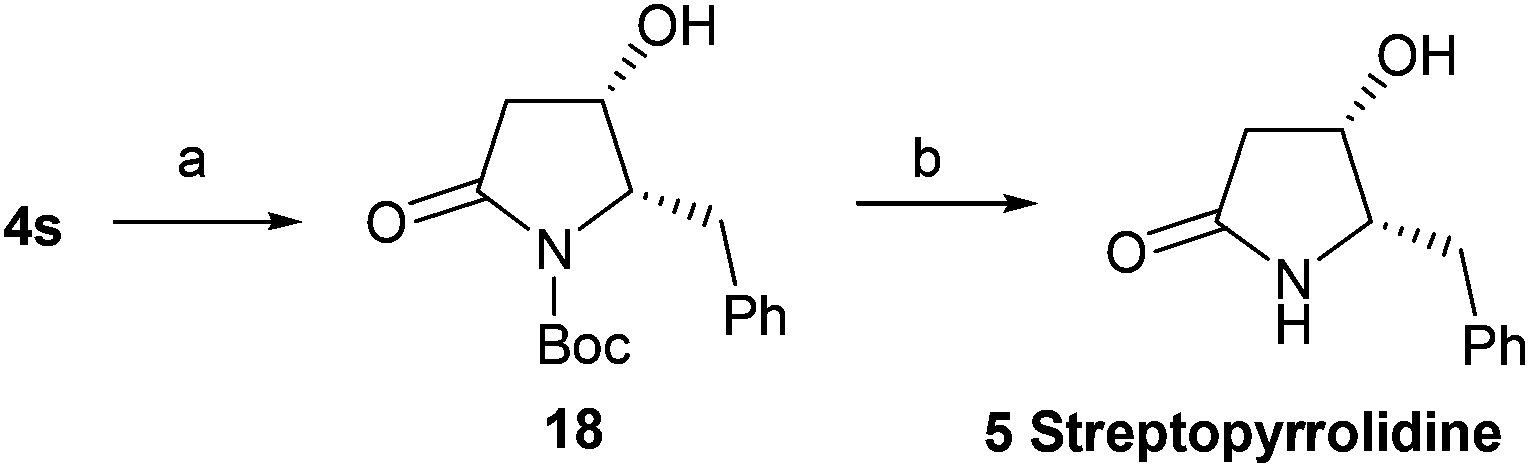
With this effective method for the preparation of trans-4-hydroxy-5-substituted 2-pyrrolidinones in hand, we decided to use it in the total syntheses of natural products. The synthesis of streptopyrrolidine 5 only requires a chiral inversion at the C-4 position of 4s. After the deprotection (tetra-n-butylammonium fluoride, TBAF) of 4s, oxidation (DMP) and subsequent reduction (NaBH4) generated 18 in 89% overall yield, which was treated with trifluoroacetic acid (TFA) to afford streptopyrrolidine 5 in 83% yield (Scheme 2) {[α]25D = −44.5 (c 0.15, MeOH); lit.9j [α]25D = −44.6 (c 0.05, MeOH); lit.11 [α]25D = −12 (c 0.05, MeOH); lit.20a [α]20D = −44 (c 1.0, MeOH); lit.20b [α]20D = −43.5 (c 1.0, MeOH)}. The spectroscopy and physical data of the synthetic streptopyrrolidine 5 were identical to the reported data.9j,11,20
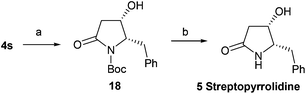 |
| | Scheme 2 Asymmetric synthesis of streptopyrrolidine 5. Reagents and conditions: (a) (1) TBAF, THF, 3 h; (2) DMP, DCM, 30 min; (3) NaBH4, MeOH, 0 °C–rt, 2 h, 89% (3 steps); (b) TFA, 0 °C–rt, 4 h, 83%. | |
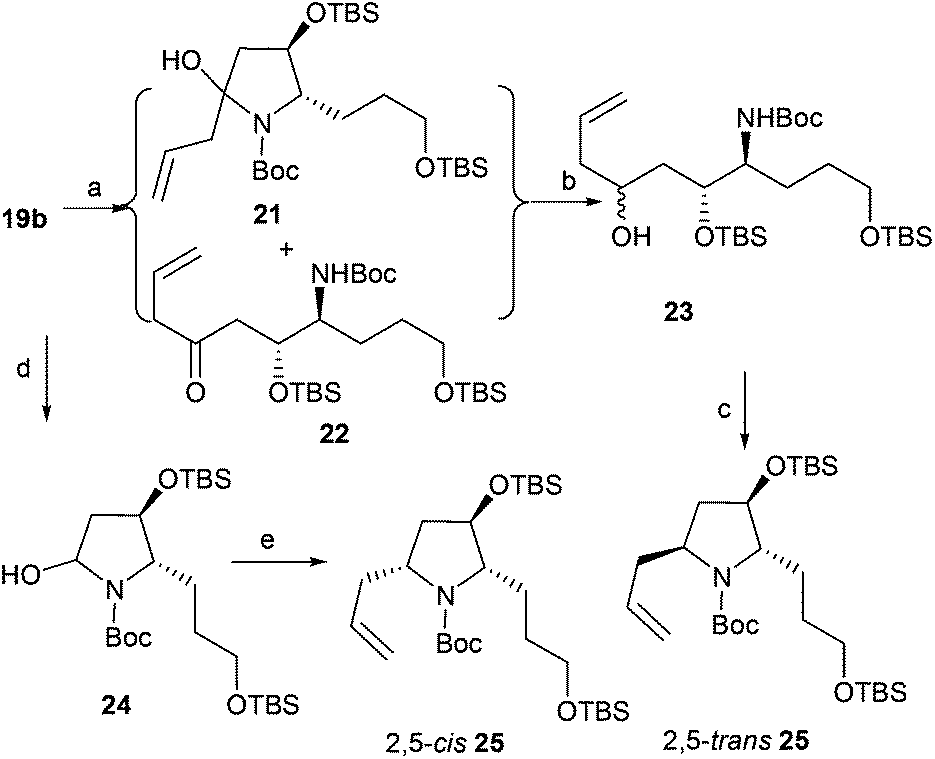
Next, we focused on the total synthesis of epohelmin A 6, a novel lanosterol synthase inhibitor that was isolated from a fungal strain FKI-0929.12a The chemical structure of 6 was revised from the originally proposed monocyclic core to a bicyclic skeleton by Snider after their asymmetric synthesis in 2005.12b,c Our retrosynthetic analysis is shown in Fig. 3, hoping to introduce the C-3 chiral center through nucleophilic addition of lactam by allylmagnesium chloride and subsequent stereoselective reduction sequence.
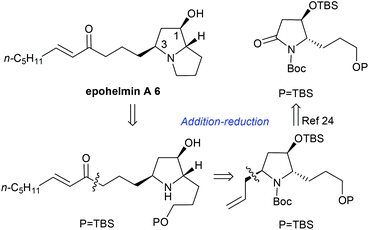 |
| | Fig. 3 Retrosynthetic analysis of epohelmin A 6. | |
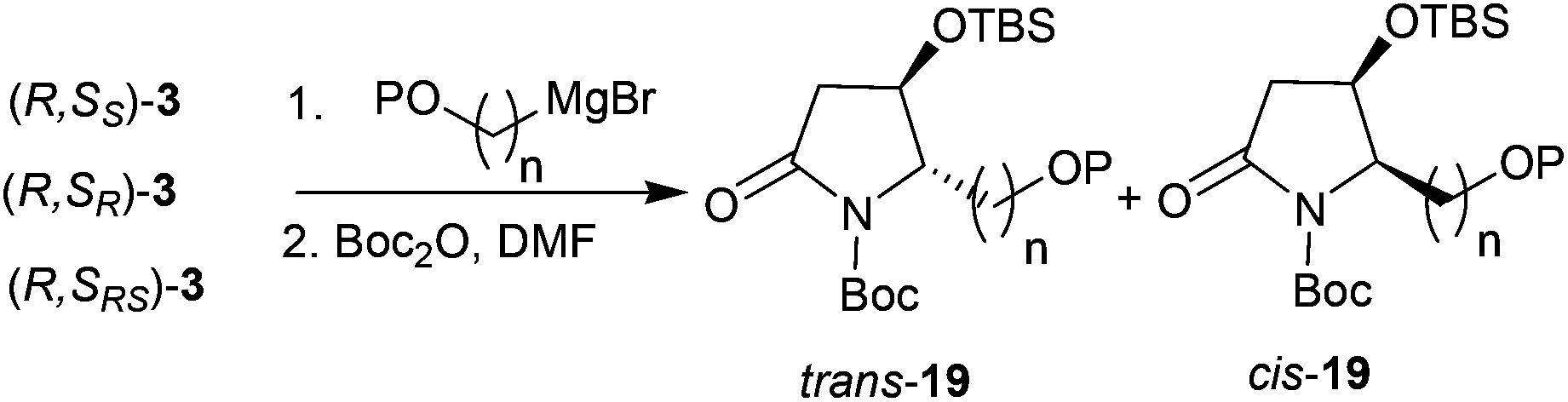
Thus, the intramolecular cascade reaction of α-chiral aldimines (R,SRS)-3 with [3-(benzyloxy)propyl]magnesium bromide was initially considered. To our disappointment, the yield of 19a and its diastereoselectivity were very low (38:62) (Table 5, entry 1). Moreover, the major imide 19a was in the cis form (Table 5, entry 1). Although several conditions including (R,SR)-3 and (R,SS)-3 were investigated, the stereochemistry was still low (Table 5, entries 2 and 3). The use of (3-[(tert-butyl)dimethylsilyloxy]propyl)magnesium bromide was also investigated, but the intramolecular tandem process did not proceed (Table 5, entry 4). To further investigate this abnormal result, different [(benzyloxy)alkyl]magnesium bromides were studied. When (R,SRS)-3 was treated with [2-(benzyloxy)ethyl]magnesium bromide, no desired product was produced (Table 5, entry 5). Using [4-(benzyloxy)butyl]magnesium bromide, the yield of desired imide 19d improved to 53%, and the ratio of diastereoselectivity of trans to cis changed to 50:50 (Table 5, entry 6). Interestingly, when (R,SRS)-3 was treated with [6-(benzyloxy)hexyl]magnesium bromide, the desired product 19e was generated in 60% yield and the diastereoselectivity returned to normal, with 90:10 ratio favoring the trans (Table 5, entry 7).
Table 5 Reactions of (benzyloxy) or (silaneoxy) Grignard reagents with 3
|
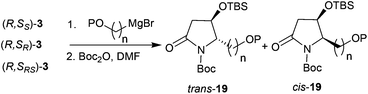
|
| Entrya |
3
|
n
|
P |
19
|
Yieldb (%) |
trans:cisc |
|
Reactions were performed with α-chiral substituted aldimines 3 (1.0 mmol), Grignard reagents (3 mL, 1.0 M in THF) in dry THF (5 mL) at −78 °C to rt overnight, crude product was treated with Boc2O (2.0 mmol), DMAP (1.0 mmol), and TEA (5.0 mmol) in DMF for 24 h.
Isolated yield.
Determined by HPLC or 1H NMR.
No reaction.
|
| 1 |
(R,SRS) |
3 |
Bn |
19a
|
32 |
38:62 |
| 2 |
(R,SR) |
3 |
Bn |
19a
|
35 |
41:59 |
| 3 |
(R,SS) |
3 |
Bn |
19a
|
29 |
35:65 |
| 4 |
(R,SRS) |
3 |
TBS |
19b
|
NRd |
— |
| 5 |
(R,SRS) |
2 |
Bn |
19c
|
NRd |
— |
| 6 |
(R,SRS) |
4 |
Bn |
19d
|
53 |
50:50 |
| 7 |
(R,SRS) |
6 |
Bn |
19e
|
60 |
90:10 |
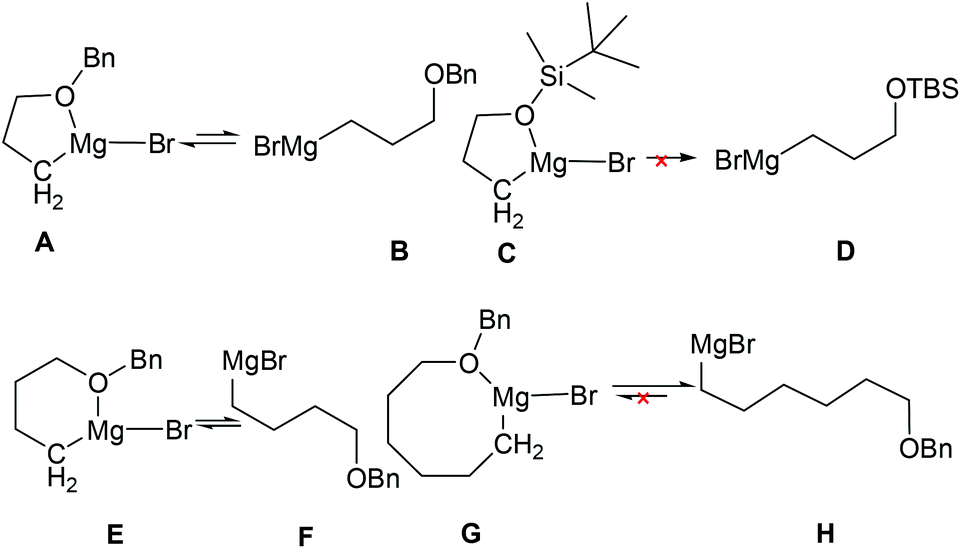
In understanding why the intramolecular cascade reaction of (R,SRS)-3 with [3-(benzyloxy)propyl]magnesium bromide gave different results, we presumed that the strong coordination of magnesium with oxygen produced the cyclic form as stable intermediates (Fig. 4, structure A)21 that weakly produced B to attack the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond. It is likely that steric hindrance caused the abnormal result. For (3-[(tert-butyl)dimethylsilyloxy]propyl)magnesium bromide D, we presume that the steric hindrance of the more stable structure C is predominant. Thus it could not attach the CN bond. However, there is balance between the coordination structure E and [4-(benzyloxy)butyl]magnesium bromide F, and the steric hindrance of E caused the cis-result. Thus, the ratio of diastereoselectivity for imine 19d is close to 50:50. For [6-(benzyloxy)hexyl]magnesium bromide H, the coordination structure G is very weak, so the intramolecular cascade reaction of (R,SRS)-3 gave the normal trans product.
N bond. It is likely that steric hindrance caused the abnormal result. For (3-[(tert-butyl)dimethylsilyloxy]propyl)magnesium bromide D, we presume that the steric hindrance of the more stable structure C is predominant. Thus it could not attach the CN bond. However, there is balance between the coordination structure E and [4-(benzyloxy)butyl]magnesium bromide F, and the steric hindrance of E caused the cis-result. Thus, the ratio of diastereoselectivity for imine 19d is close to 50:50. For [6-(benzyloxy)hexyl]magnesium bromide H, the coordination structure G is very weak, so the intramolecular cascade reaction of (R,SRS)-3 gave the normal trans product.
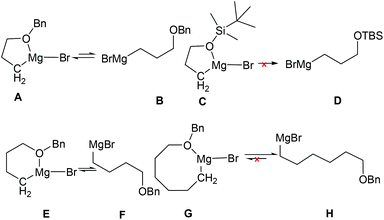 |
| | Fig. 4 The proposed coordination structures of magnesium with oxygen. | |
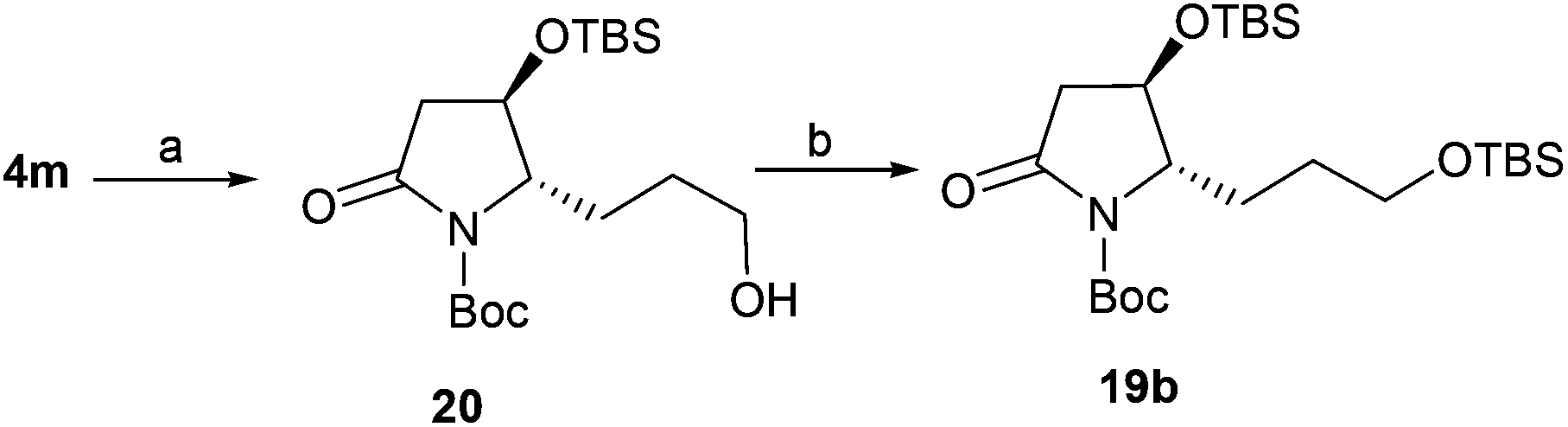
To obtain 19b, lactam 4m was selected as a key intermediate for the synthesis of epohelmin A 6. Thus, the dihydroxylation of alkene 4m with 4-methylmorpholine 4-oxide (NMO) in the presence of 0.1 equivalents of K2Os2O2(OH)422 and continuous oxidation with NaIO423 as well as subsequent reduction with NaBH4 gave desired alcohol 20 in 87% overall yield (Scheme 3). The hydroxyl group in 20 was then converted to TBS ether in 93% yield.
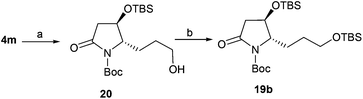 |
| | Scheme 3 Synthesis of 19b. Reagents and conditions: (a) (1) NMO, K2OsO4, t-BuOH/H2O, overnight; (2) NaIO4, THF/H2O, 1.5 h; (3) NaBH4, MeOH, 0 °C, 87% (3 steps); (b) TBSCl, DMAP, imidazole, DMF, 24 h, 93%. | |
Imine 19b was treated with a solution of allylmagnesium chloride in dichloromethane at −20 °C; additive product 21 and the tautomeric amidoketone 22 were generated24 (Scheme 4). Without further purification, the crude mixture of compounds 21 and 22 was treated with sodium borohydride (ethanol/NaBH4) in the presence of cerous chloride to afford the crude alcohol 23, which was directly treated with methanesulfonyl chloride and potassium tert-butoxide to give 25 in 70% overall yield with diastereoselectivity (dr = 50:50). Methanol was also used as a solvent, but the result remained the same (dr = 50:50). To obtain diastereoselectivity in 25, the diastereoselective nucleophilic addition of organic boronic ester to N-acyliminium ions24b was investigated. Reduction of 19b with lithium triethylhydridoborate gave N,O-acetals 24 in quantitative yield, which was directly treated with 2-allyl-4,4,5,5-tetramethyl-1,3,2-dioxaborolane to afford desired product 2,5-cis-25 in 69% overall yield with high diastereoselectivity (dr > 99:1). The stereochemistry of 25 prepared by the addition–reduction–cyclization process was assigned as the 2,5-trans form by comparing data with 2,5-cis-25.24b
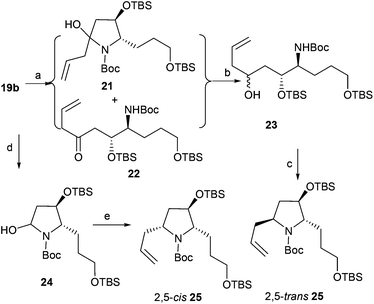 |
| | Scheme 4 Asymmetric allylation of 19b. Reagents and conditions: (a) allylmagnesium chloride, DCM; (b) NaBH4, CeCl3, EtOH; (c) (1) MsCl, TEA, DCM; (2) t-BuOK, THF, 70% (3 steps); (d) LiBEt3H, THF, −78 °C; (e) 2-allyl-4,4,5,5-tetramethyl-1,3,2-dioxaborolane, BF3·Et2O, −78 °C to −40 °C, DCM, 69% (2 steps from 19b). | |
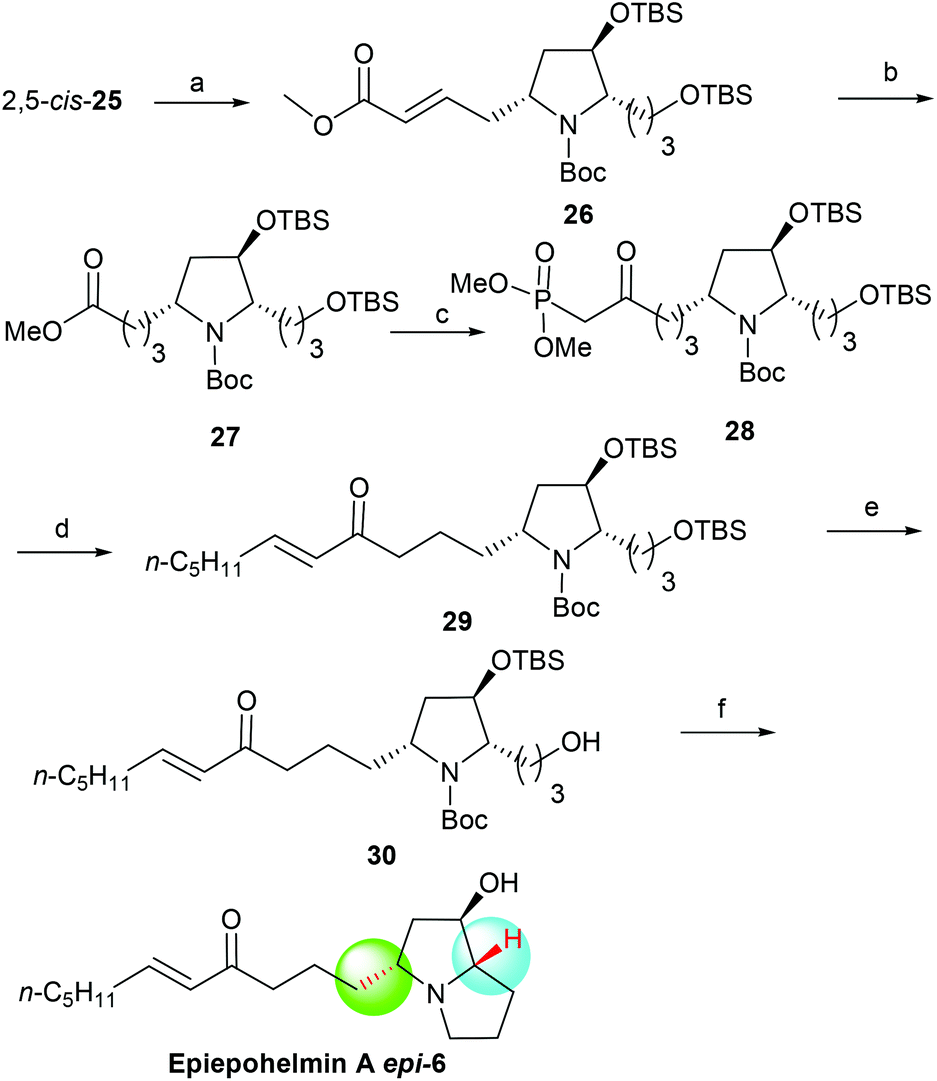
With the 2,5-cis-25 in hand, the asymmetric synthesis of 3-epi-epohelmin A (3-epi-6) was considered as shown in Scheme 5. The carbon chain extension of 2,5-cis-25 through Grubbs’ second-generation catalyst25 successfully afforded olefin 26 in 95% yield with excellent selectivity (E:Z = 95:5). Reduction (Pd/C, H2) of 26 gave ester 27 in 92% yield. Compound 27 was treated with a large excess of LiCH2PO(OMe)2 in THF from −78 °C to −50 °C to obtain keto phosphonate 28 in 97% isolated yield.12b Treatment of 28 with NaH in THF, followed by the addition of hexanal gave enone 29 in 88% yield with good selectivity (E:Z = 98:2).12b Selective deprotection of 29 with CSA in a mixture of DCM and methanol at −50 °C for 8 h gave alcohol 30 in 93% yield. Finally, mesylation of compound 30 with methanesulfonyl chloride in the presence of triethylamine and subsequent treatment with triethylsilyl triflate (TESOTf)/2,6-lutidine generated the bicyclic product, which was subjected to desilylation with HCl/methanol in one pot, affording 3-epi-epohelmin A (3-epi-6) in 49% yield {[α]25D = +7.8 (c 0.80, CHCl3)}. The structure of the synthetic 3-epi-6 was further confirmed by the spectroscopy and physical data.
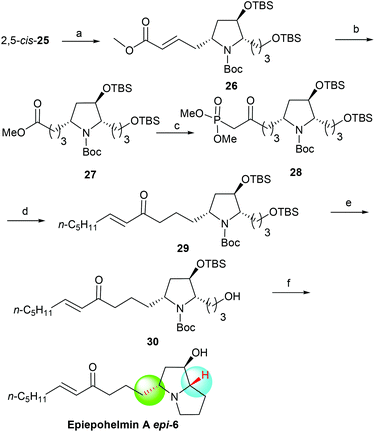 |
| | Scheme 5 Asymmetric synthesis of 3-epi-epohelmin A (3-epi-6). Reagents and conditions: (a) methyl acrylate, Grubbs 2nd generation catalyst, DCM, reflux, 7 h, 95%; (b) Pd/C, H2, MeOH, 92%; (c) dimethyl methylphosphonate, n-BuLi, THF, −78 °C, 1 h, then (5R)-27, −50 °C, 2 h, 97%; (d) NaH, (5R)-28, THF, 1 h, then caproaldehyde, 0 °C, 3 h, 88%; (e) CSA, DCM/MeOH, −50 °C, 8 h, 93%; (f) (1) MsCl, TEA, DCM, 100%; (2) 2,6-lutidine, TESOTf, DCM, −78 °C–rt, overnight; (3) HCl/MeOH, 24 h, 49% (2 steps). | |
Conclusions
We established a convenient one-pot method for highly diastereoselective synthesis of trans-4-hydroxy-5-substituted 2-pyrrolidinones 4a–s, 14a–e, and 17a,b by an intramolecular cascade process of (R,SRS)-3 with alkyl, aryl, alkynyl, and alkenyl Grignard reagents. For the reaction of (R,SRS)-3 with alkyl, aryl, and alkenyl Grignard reagents, the stereochemistry at the C-5 stereogenic center of the trans-4-hydroxy-5-substituted 2-pyrrolidinones was solely controlled by α-alkoxy substitution. In contrast, in reactions of (R,SRS)-3 with alkynyl Grignard reagents, the stereochemistry at the C-5 stereogenic center was controlled by coordination of the α-alkoxy substitution and the stereochemistry of the sulfinamide. The synthetic application of this methodology was demonstrated by the asymmetric syntheses of streptopyrrolidine 5 and 3-epi-epohelmin A (3-epi-6).
Experimental section
General
THF was distilled from sodium/benzophenone. The reactions were monitored by thin layer chromatography (TLC) on glass plates coated with silica gel with a fluorescent indicator. Flash chromatography was performed on silica gel (300–400) with petroleum/EtOAc as the eluent. Optical rotations were measured on a polarimeter with a sodium lamp. HRMS were measured on a LCMS-IT-TOF apparatus. IR spectra were recorded using film on a Fourier Transform Infrared Spectrometer. NMR spectra were recorded at 400 MHz or 500 MHz, and chemical shifts are reported in δ (ppm) referenced to an internal TMS standard for 1H NMR and CDCl3 (77.0 ppm) for 13C NMR.
(R)-Methyl 3-(tert-butyldimethylsilyloxy)-4-hydroxybutanoate 10.
A solution of 9 (40.0 g, 110.42 mmol) and CSA (20.9 g, 88.34 mmol) was stirred in DCM (200 mL) and MeOH (200 mL) at −40 °C for 8 h. Then a solution of TEA (15.3 mL, 110.42 mmol) was added and the mixture was warmed to room temperature. After being concentrated, the residue was purified by flash chromatography on a silica gel column (PE/EA = 6/1) to give alcohol 10 (17.8 g, 65%) as a colorless oil. [α]25D = +12.8 (c 1.02, CHCl3); IR (film): νmax 3462, 2954, 2930, 2887, 2858, 1740, 1438, 1256, 1169, 1115, 1071, 838, 779 cm−1; 1H NMR (400 MHz, CDCl3) δ 4.24–4.16 (m, 1H), 3.67 (s, 3H), 3.63–3.57 (m, 1H), 3.56–3.50 (m, 1H), 2.60–2.50 (m, 2H), 2.03 (dd, J = 7.6, 5.2 Hz, 1H), 0.87 (s, 9H), 0.09 (s, 3H), 0.07 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3) δ 171.9, 69.7, 66.2, 51.6, 39.2, 25.7, 18.0, −4.7, −4.9 ppm; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C11H24O4SiNa: 271.1342, found: 271.1354.
(E,3R,SRS)-Methyl 3-(tert-butyldimethylsilyloxy)-4-(2-methylpropan-2-ylsulfinamido)butanoate 3.
Then the above alcohol 10 was dissolved in DCM (200 mL) and treated with freshly prepared DMP (36.5 g, 86.14 mmol) at room temperature for 30 min. The mixture was quenched carefully with a saturated solution of NaHCO3 and Na2S2O3. The resulted mixture was separated and the aqueous layer was extracted with DCM three times. The combined organic layers were washed with brine, dried, filtered and concentrated to give crude aldehyde 11, which was dissolved in DCM (200 mL). Then 2-methyl-2-propanesulfinamide (8.7 g, 71.78 mmol), cupric sulphate anhydrous (22.9 g, 143.56 mmol) and PPTS (0.9 g, 3.59 mmol) were added in one portion. After being stirred for 24 h, the reaction was filtered and concentrated to give the crude product, which was purified by flash chromatography on a silica gel column (PE/EA = 10/1) to give 3 (21.1 g, 2 steps for 84%) as a colorless oil. For the mixture: IR (film): νmax 2955, 2930, 2898, 2858, 1742, 1624, 1473, 1462, 1437, 1364, 1256, 1173, 1090, 1005, 838, 780 cm−1; 1H NMR (400 MHz, CDCl3) δ 8.06–8.02 (m, 0.33H), δ 7.96–7.94 (m, 0.67H), 4.90–4.84 (m, 0.67H), 4.58–4.54 (m, 0.33H), 3.68–3.67 (m, 1H), 3.65–3.63 (m, 2H), 2.98–2.85 (m, 0.66H), 2.78–2.71 (m, 0.67H), 2.62–2.54 (m, 0.67H), 1.16–1.14 (m, 6H), 1.14–1.12 (m, 3H), 0.86–0.82 (m, 9H), 0.07–0.02 (m, 6H) ppm; 13C NMR (100 MHz, CDCl3) δ 172.6, 170.7, 169.4, 165.7, 71.2, 69.3, 57.0, 56.8, 52.0, 51.8, 40.9, 40.3, 25.6, 22.3, 22.2, 18.2, 18.0, −4.5, −4.9, −5.1, −5.4 ppm; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C15H32NO4SSi: 350.1821, found: 350.1821.
General procedure for synthesis of 4a–s, 14a–e and 17a–b.
Compound 3 (350 mg, 1.00 mmol) was dissolved in anhydrous THF (5 mL) and cooled to −78 °C. Then a solution of Grignard reagent (3 mL, 1 M in THF) was slowly added. After stirring for 3 h, the mixture was warmed to room temperature and stirred overnight. The reaction was quenched with a saturated NH4Cl aqueous solution and extracted with EtOAc (30 mL × 3). The combined organic layers were washed with brine, dried, filtered and concentrated to give crude amide, which was directly dissolved in dry DMF (4 mL), then TEA (0.70 mL, 5.00 mmol), Boc2O (0.46 mL, 2 mmol) and DMAP (122 mg, 1.00 mmol) were added. After stirring for 24 h, the reaction was diluted with water and extracted with EtOAc for three times. The combined organic layers were washed with water and brine two times respectively. The organic layer was dried, filtered and concentrated. Then the residue was purified by flash chromatography on a silica gel column (PE/EA = 15/1) to give imide 4a–s, 14a–e and 17a–b.
(2S,3R)-tert-Butyl 3-(tert-butyldimethylsilyloxy)-5-oxo-2-phenylpyrrolidine-1-carboxylate 4a.
White solid (277 mg, 71%), m.p. 112–113 °C. [α]25D = −5.4 (c 1.40, CHCl3); IR (film): νmax 2948, 2931, 2855, 1794, 1458, 1367, 1306, 1255, 1151, 1101, 839, 780 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.41–7.30 (m, 3H), 7.22–7.18 (m, 2H), 5.00–4.95 (m, 1H), 4.12 (ddd, J = 5.6, 2.4, 1.6 Hz, 1H), 2.87 (dd, J = 17.2, 5.6 Hz, 1H), 2.43 (dd, J = 17.2, 1.6 Hz, 1H), 1.32 (s, 9H), 0.91 (s, 9H), 0.08 (s, 3H), 0.06 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3) δ 172.9, 149.6, 139.1, 129.0, 128.0, 125.2, 83.0, 72.0, 71.4, 41.0, 27.7, 25.7, 18.0, −4.7, −4.8 ppm; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C21H33NO4SiNa: 414.2077, found: 414.2066.
(2S,3R)-tert-Butyl 3-(tert-butyldimethylsilyloxy)-5-oxo-2-o-tolylpyrrolidine-1-carboxylate 4b.
Pale yellow oil (251 mg, 62%). [α]25D = −8.2 (c 2.50, CHCl3); IR (film): νmax 2959, 2931, 2849, 1789, 1753, 1721, 1463, 1370, 1307, 1249, 1153, 1079, 838 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.24–7.16 (m, 3H), 7.06–7.01 (m, 1H), 5.23–5.21 (m, 1H), 4.07 (ddd, J = 5.6, 2.4, 1.6 Hz, 1H), 2.87 (dd, J = 17.2, 5.6 Hz, 1H), 2.45–2.39 (m, 4H), 1.28 (s, 9H), 0.90 (s, 9H), 0.04 (s, 3H), 0.03 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3) δ 173.0, 149.4, 137.1, 134.9, 130.8, 127.7, 126.6, 123.2, 82.8, 71.1, 67.9, 41.1, 27.6, 25.6, 19.3, 17.8, −4.6, −4.9 ppm; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C22H35NO4SiNa: 428.2233, found: 428.2231.
(2S,3R)-tert-Butyl 3-(tert-butyldimethylsilyloxy)-5-oxo-2-m-tolylpyrrolidine-1-carboxylate 4c.
Pale yellow oil (296 mg, 73%). [α]25D = −2.4 (c 2.78, CHCl3); IR (film): νmax 2959, 2931, 2849, 1789, 1753, 1715, 1359, 1307, 1154, 1079, 838, 778 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.24–7.17 (m, 1H), 7.08–7.04 (m, 1H), 6.95–6.90 (m, 2H), 4.90–4.87 (m, 1H), 4.05 (ddd, J = 5.6, 2.4, 1.6 Hz, 1H), 2.81 (dd, J = 17.6, 5.6 Hz, 1H), 2.35 (dd, J = 17.6, 1.6 Hz, 1H), 2.29 (s, 3H), 1.26 (s, 9H), 0.84 (s, 9H), 0.03 (s, 3H), 0.00 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3) δ 173.0, 149.6, 138.9, 138.7, 128.9, 128.7, 125.6, 122.3, 82.9, 72.0, 71.4, 41.1, 27.7, 25.7, 21.4, 18.0, −4.7, −4.8 ppm; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C22H35NO4SiNa: 428.2233, found: 428.2233.
(2S,3R)-tert-Butyl 3-(tert-butyldimethylsilyloxy)-5-oxo-2-p-tolylpyrrolidine-1-carboxylate 4d.
White solid (276 mg, 68%), m.p. 98–99 °C. [α]25D = −4.6 (c 1.43, CHCl3); IR (film): νmax 2953, 2932, 2855, 1789, 1726, 1370, 1307, 1153, 1068, 921, 838, 777 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.19–7.17 (m, 2H), 7.08–7.06 (m, 2H), 4.97–4.93 (m, 1H), 4.09 (ddd, J = 5.6, 2.0, 1.2 Hz, 1H), 2.86 (dd, J = 17.6, 5.6 Hz, 1H), 2.40 (ddd, J = 17.6, 2.0, 0.8 Hz, 1H), 2.36 (s, 3H), 1.33 (s, 9H), 0.90 (s, 9H), 0.09 (s, 3H), 0.06 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3) δ 173.0, 149.7, 137.7, 136.0, 129.6, 125.1, 82.9, 72.1, 71.3, 41.0, 27.7, 25.7, 21.1, 18.0, −4.7, −4.8 ppm; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C22H35NO4SiNa: 428.2233, found: 428.2222.
(2S,3R)-tert-Butyl 3-(tert-butyldimethylsilyloxy)-2-(4-methoxyphenyl)-5-oxopyrrolidine-1-carboxylate 4e.
Pale yellow oil (312 mg, 74%). [α]25D = −5.7 (c 1.66, CHCl3); IR (film): νmax 2953, 2932, 2849, 1787, 1715, 1512, 1364, 1306, 1251, 1153, 1079, 838 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.03–7.09 (m, 2H), 6.93–6.89 (m, 2H), 4.94–4.92 (m, 1H), 4.08 (ddd, J = 5.6, 2.0, 1.2 Hz, 1H), 3.82 (s, 3H), 2.87 (dd, J = 17.6, 5.6 Hz, 1H), 2.41 (dd, J = 17.6, 1.6 Hz, 1H), 1.34 (s, 9H), 0.90 (s, 9H), 0.08 (s, 3H), 0.06 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3) δ 172.9, 159.3, 149.6, 131.1, 126.4, 114.3, 82.9, 72.1, 70.1, 55.3, 41.0, 27.8, 25.7, 18.0, −4.7, −4.8 ppm; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C22H35NO5SiNa: 444.2182, found: 444.2172.
(2S,3R)-tert-Butyl 3-(tert-butyldimethylsilyloxy)-2-(3-fluorophenyl)-5-oxopyrrolidine-1-carboxylate 4f.
White solid (327 mg, 80%), m.p. 51–53 °C. [α]25D = −6.0 (c 1.9, CHCl3); IR (film): νmax 2954, 2931, 2858, 1790, 1758, 1721, 1589, 1453, 1367, 1305, 1258, 1153, 1082, 919, 839, 779 cm−1; 19F NMR (376 MHz, CDCl3) δ −111.82 ppm; 1H NMR (400 MHz, CDCl3) δ 7.40–7.33 (m, 1H), 7.05–6.98 (m, 2H), 6.93–6.88 (m, 1H), 4.97–4.93 (m, 1H), 4.10 (ddd, J = 5.6, 2.8, 2.0 Hz, 1H), 2.86 (dd, J = 17.6, 5.6 Hz, 1H), 2.44 (dd, J = 17.6, 2.4 Hz, 1H), 1.34 (s, 9H), 0.90 (s, 9H), 0.07 (s, 3H), 0.06 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3) δ 172.4, 163.2 (d, J = 246.1 Hz), 149.4, 141.9 (d, J = 6.7 Hz), 130.7 (d, J = 8.2 Hz), 120.7 (d, J = 2.2 Hz), 114.9 (d, J = 21.0 Hz), 112.4 (d, J = 22.2 Hz), 83.3, 71.9, 70.9, 41.0, 27.7, 25.6, 18.0, −4.5, −4.8, −5.1 ppm; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C21H32FNO4SiNa: 432.1982, found: 432.1985.
(2S,3R)-tert-Butyl 3-(tert-butyldimethylsilyloxy)-2-(4-fluorophenyl)-5-oxopyrrolidine-1-carboxylate 4g.
White solid (331 mg, 81%), m.p. 111–112 °C. [α]25D = −6.9 (c 0.78, CHCl3); IR (film): νmax 2948, 2926, 2849, 1789, 1715, 1518, 1370, 1306, 1249, 1151, 1085, 839 cm−1; 19F NMR (376 MHz, CDCl3) δ −114.11 ppm; 1H NMR (400 MHz, CDCl3) δ 7.21–7.15 (m, 2H), 7.12–7.06 (m, 2H), 4.95–4.92 (m, 1H), 4.08 (ddd, J = 5.6, 2.8, 2.0 Hz, 1H), 2.86 (dd, J = 17.6, 6.0 Hz, 1H), 2.45 (dd, J = 17.6, 2.4 Hz, 1H), 1.34 (s, 9H), 0.90 (s, 9H), 0.06 (s, 3H), 0.05 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3) δ 172.5, 162.3 (d, J = 245.2 Hz), 149.4, 135.0 (d, J = 2.8 Hz), 126.9 (d, J = 8.1 Hz), 116.0 (d, J = 21.5 Hz), 83.2, 72.0, 70.7, 41.0, 27.7, 25.6, 18.0, −4.5, −4.8, −5.1 ppm; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C21H32FNO4SiNa: 432.1982, found: 432.1977.
(2S,3R)-tert-Butyl 3-(tert-butyldimethylsilyloxy)-5-oxo-2-(3-(trifluoromethyl)phenyl)pyrrolidine-1-carboxylate 4h.
Pale yellow oil (271 mg, 59%). [α]25D = −4.1 (c 0.92, CHCl3); IR (film): νmax 2948, 2931, 2866, 1801, 1452, 1364, 1333, 1255, 1167, 1130, 1096, 893, 839, 780 cm−1; 19F NMR (376 MHz, CDCl3) δ −62.78 ppm; 1H NMR (400 MHz, CDCl3) δ 7.63–7.38 (m, 4H), 4.97 (d, J = 2.4 Hz, 1H), 4.11 (ddd, J = 6.4, 3.2, 2.8 Hz, 1H), 2.86 (dd, J = 17.2, 6.4 Hz, 1H), 2.49 (dd, J = 17.6, 3.2 Hz, 1H), 1.31 (s, 9H), 0.89 (s, 9H), 0.04 (s, 3H), 0.03 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3) δ 172.2, 149.3, 140.5, 131.5 (d, J = 43.1, 21.6 Hz), 129.6, 128.5, 124.8 (d, J = 23.0 Hz), 123.8 (d, J = 361.0, 180.4 Hz), 122.3 (d, J = 22.0 Hz), 83.5, 72.0, 70.8, 41.0, 27.7, 25.6, 17.9, −4.8, −4.9 ppm; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C22H32F3NO4SiNa: 482.1950, found: 482.1951.
(2S,3R)-tert-Butyl 3-(tert-butyldimethylsilyloxy)-2-(4-phenyl)-5-oxopyrrolidine-1-carboxylate 4i.
Colorless oil (378 mg, 81%). [α]25D = −5.8 (c 2.75, CHCl3); IR (film): νmax 2953, 2937, 2849, 1788, 1721, 1359, 1306, 1255, 1152, 1079, 838 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.64–7.61 (m, 4H), 7.49–7.45 (m, 2H), 7.40–7.36 (m, 1H), 7.27–7.25 (m, 2H), 5.05–5.03 (m, 1H), 4.17 (ddd, J = 5.2, 3.2, 1.2 Hz, 1H), 2.92 (dd, J = 17.6, 5.6 Hz, 1H), 2.40 (dd, J = 17.6, 1.6, Hz, 1H), 1.36 (s, 9H), 0.93 (s, 9H), 0.12 (s, 3H), 0.09 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3) δ 172.8, 149.7, 140.9, 140.3, 138.0, 128.9, 127.7, 127.6, 127.0, 125.6, 83.1, 72.0, 71.2, 41.1, 27.8, 25.7, 18.0, −4.7, −4.8 ppm; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C27H37NO4SiNa: 490.2390, found: 490.2381.
(2S,3R)-tert-Butyl 3-(tert-butyldimethylsilyloxy)-2-(naphthalen-1-yl)-5-oxopyrrolidine-1-carboxylate 4j.
Colorless oil (260 mg, 59%). [α]25D = +42.2 (c 1.80, CHCl3); IR (film): νmax 2953, 2932, 2860, 1788, 1759, 1726, 1370, 1308, 1153, 1074, 921, 838, 778 cm−1; 1H NMR (400 MHz, CDCl3) δ 8.07–8.05 (m, 1H), 7.95–7.92 (m, 1H), 7.84–7.82 (m, 1H), 7.62–7.54 (m, 2H), 7.47–7.43 (m, 1H), 7.28–7.25 (m, 1H), 5.87–5.85 (m, 1H), 4.24 (ddd, J = 5.6, 1.6, 1.2 Hz, 1H), 2.85 (dd, J = 17.6, 5.2 Hz, 1H), 2.43 (d, J = 17.6 Hz, 1H), 1.26 (s, 9H), 0.96 (s, 9H), 0.10 (s, 3H), 0.04 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3) δ 173.2, 149.7, 134.3, 134.0, 130.2, 129.1, 128.5, 126.6, 126.1, 125.3, 122.6, 120.8, 83.0, 70.8, 68.2, 41.3, 27.7, 25.7, 17.9, −4.5, −4.7 ppm; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C25H35NO4SiNa: 464.2233, found: 464.2223.
(2S,3R)-tert-Butyl 3-(tert-butyldimethylsilyloxy)-2-(naphthalen-2-yl)-5-oxopyrrolidine-1-carboxylate 4k.
White solid (309 mg, 70%), m.p. 116–117 °C. [α]25D = −2.6 (c 0.44, CHCl3); IR (film): νmax 2953, 2932, 2866, 1789, 1748, 1715, 1468, 1370, 1306, 1255, 1153, 1068, 838, 779 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.91–7.82 (m, 3H), 7.63 (s, 1H), 7.56–7.50 (m, 2H), 7.35–7.33 (m, 1H), 5.17–5.15 (m, 1H), 4.20 (ddd, J = 5.2, 2.0, 1.2 Hz, 1H), 2.94 (dd, J = 17.4, 5.6 Hz, 1H), 2.47 (dd, J = 17.4, 1.6 Hz, 1H), 1.31 (s, 3H), 0.93 (s, 9H), 0.11 (s, 3H), 0.08 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3) δ 173.0, 149.7, 136.3, 133.3, 133.0, 129.1, 127.9, 127.7, 126.7, 126.3, 123.6, 123.3, 83.1, 71.8, 71.5, 41.1, 27.7, 25.7, 18.0, −4.7, −4.7 ppm; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C25H35NO4SiNa: 464.2233, found: 464.2231.
(2S,3R)-tert-Butyl 3-(tert-butyldimethylsilyloxy)-2-ethyl-5-oxopyrrolidine-1-carboxylate 4l.
Colorless oil (223 mg, 65%). [α]25D = +28.6 (c 1.28, CHCl3); IR (film): νmax 2948, 2926, 2866, 1797, 1753, 1714, 1463, 1370, 1309, 1249, 1156, 1074, 926, 837, 777 cm−1; 1H NMR (400 MHz, CDCl3) δ 4.00 (d, J = 5.2 Hz, 1H), 3.79 (dd, J = 9.6, 3.6 Hz, 1H), 2.68 (dd, J = 17.6, 5.2 Hz, 1H), 2.27 (d, J = 17.6 Hz, 1H), 1.74–1.61 (m, 1H), 1.46 (s, 9H), 1.39–1.28 (m, 1H), 0.94–0.88 (m, 3H), 0.80 (s, 9H), 0.01 (s, 3H), 0.00 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3) δ 172.7, 150.1, 82.7, 69.2, 67.9, 41.8, 28.0, 25.7, 24.9, 17.9, 10.3, −4.6, −4.7 ppm; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C17H33NO4SiNa: 366.2077, found: 366.2067.
(2S,3R)-tert-Butyl 2-(but-3-enyl)-3-(tert-butyldimethylsilyloxy)-5-oxopyrrolidine-1-carboxylate 4m.
White solid (255 mg, 69%), m.p. 51–53 °C. [α]25D = +30.0 (c 1.00, CHCl3); IR (film): νmax 2955, 2930, 2857, 1787, 1754, 1716, 1468, 1369, 1311, 1258, 1155, 1081, 915, 837 cm−1; 1H NMR (400 MHz, CDCl3) δ 5.86–5.75 (m, 1H), 5.12–5.00 (m, 2H), 4.10–4.06 (m, 1H), 3.98–3.92 (m, 1H), 2.82–2.74 (m, 1H), 2.38–2.30 (m, 1H), 2.23–2.05 (m, 2H), 1.84–1.75 (m, 1H), 1.55–1.52 (m, 9H), 1.51–1.43 (m, 1H), 0.88–0.85 (m, 9H), 0.09–0.06 (m, 6H) ppm; 13C NMR (100 MHz, CDCl3) δ 172.4, 149.8, 136.7, 115.6, 82.7, 68.0, 67.2, 41.5, 30.9, 29.9, 27.9, 25.5, 17.7, −4.8, −4.9 ppm; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C19H36NO4Si: 370.2414, found: 370.2408.
(2S,3R)-tert-Butyl 3-(tert-butyldimethylsilyloxy)-2-isopropyl-5-oxopyrrolidine-1-carboxylate 4n.
Colorless oil (186 mg, 52%). {[α]25D = +31.8 (c 1.82, CHCl3), lit.9j [α]25D = +45.17 (c = 1 in CHCl3)}; IR (film): νmax 2964, 2926, 2860, 1787, 1752, 1717, 1474, 1370, 1306, 1157, 1068, 915, 837, 777 cm−1; 1H NMR (400 MHz, CDCl3) δ 4.14 (d, J = 5.2 Hz, 1H), 3.89 (d, J = 5.6 Hz, 1H), 2.72 (dd, J = 18.0, 5.6 Hz, 1H), 2.34 (d, J = 17.6 Hz, 1H), 2.06–1.95 (m, 1H), 1.53 (s, 9H), 1.01 (d, J = 6.8 Hz, 2H), 0.90–0.86 (m, 12H), 0.09 (s, 3H), 0.08 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3) δ 173.1, 150.3, 82.8, 72.9, 65.9, 43.0, 29.8, 29.7, 28.0, 25.6, 19.3, 17.9, 17.6, −4.6, −4.7 ppm; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C18H35NO4SiNa: 380.2233, found: 380.2241.
(2S,3R)-tert-Butyl 3-(tert-butyldimethylsilyloxy)-2-isobutyl-5-oxopyrrolidine-1-carboxylate 4o.
Colorless oil (226 mg, 61%). {[α]25D = +36.8 (c 1.00, CHCl3), lit.9j [α]25D = +36.06 (c 1.02, CHCl3); lit.10c [α]25D = −35.9 (c 2.3, CHCl3)}; IR (film): νmax 2957, 2931, 2855, 1787, 1754, 1716, 1463, 1368, 1312, 1257, 1157, 1078, 915, 836, 776 cm−1; 1H NMR (400 MHz, CDCl3) δ 4.06 (d, J = 4.8 Hz, 1H), 4.01 (dd, J = 10.8, 3.2 Hz, 1H), 2.79 (dd, J = 17.6, 5.2 Hz, 1H), 2.34 (d, J = 17.6 Hz, 1H), 1.71–1.62 (m, 1H), 1.55 (s, 9H), 1.53–1.45 (m, 1H), 1.28 (ddd, J = 14.8, 10.8, 4.0 Hz, 1H), 1.04–0.96 (m, 6H), 0.88 (s, 9H), 0.09 (s, 3H), 0.08 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3) δ 172.3, 149.9, 82.8, 68.5, 66.4, 41.5, 41.1, 28.1, 25.6, 25.3, 23.8, 21.7, 17.9, −4.7, −4.8 ppm; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C19H37NO4SiNa: 394.2390, found: 394.2390.
(2S,3R)-tert-Butyl 3-(tert-butyldimethylsilyloxy)-5-oxo-2-pentylpyrrolidine-1-carboxylate 4p.
Colorless oil (277 mg, 72%). [α]25D = +20.7 (c 1.55, CHCl3); IR (film): νmax 2953, 2926, 2855, 1787, 1748, 1704, 1463, 1370, 1310, 1155, 1074, 836, 777 cm−1; 1H NMR (400 MHz, CDCl3) δ 4.09–4.05 (m, 1H), 3.95–3.89 (m, 1H), 2.76 (dd, J = 17.6, 5.2 Hz, 1H), 2.34 (d, J = 17.6 Hz, 1H), 1.71–1.64 (m, 1H), 1.56–1.52 (m, 9H), 1.43–1.26 (m, 7H), 0.95–0.85 (m, 12H), 0.10–0.05 (m, 6H) ppm; 13C NMR (100 MHz, CDCl3) δ 172.7, 150.1, 82.7, 68.3, 68.0, 41.7, 32.0, 31.6, 28.0, 25.7, 25.6, 22.5, 17.9, 13.9, −4.6, −4.7 ppm; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C20H39NO4SiNa: 408.2546, found: 408.2555.
(2S,3R)-tert-Butyl 3-(tert-butyldimethylsilyloxy)-2-cyclopropyl-5-oxopyrrolidine-1-carboxylate 4q.
Colorless oil (241 mg, 68%). [α]25D = +22.5 (c 1.95, CHCl3); IR (film): νmax 2955, 2930, 2849, 1787, 1754, 1717, 1463, 1368, 1307, 1255, 1156, 1081, 930, 838, 777 cm−1; 1H NMR (400 MHz, CDCl3) δ 4.15 (d, J = 4.8 Hz, 1H), 3.40 (d, J = 9.2 Hz, 1H), 2.76 (dd, J = 17.2, 4.8 Hz, 1H), 2.27 (d, J = 17.2 Hz, 1H), 1.46 (s, 9H), 0.79 (s, 9H), 0.72–0.62 (m, 1H), 0.60–0.44 (m, 3H), 0.24–0.14 (m, 1H), 0.00 (s, 3H), −0.01 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3) δ 172.8, 150.3, 82.7, 71.4, 70.3, 41.8, 27.9, 25.6, 17.9, 13.6, 4.5, 1.8, −4.8, −4.9 ppm; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C18H33NO4SiNa: 378.2077, found: 378.2083.
(2S,3R)-tert-Butyl 3-(tert-butyldimethylsilyloxy)-2-cyclohexyl-5-oxopyrrolidine-1-carboxylate 4r.
Colorless oil (262 mg, 66%) {[α]25D = +46.2 (c 3.33, CHCl3), lit.9j [α]25D = +40.73 (c 0.88, CHCl3)}; IR (film): νmax 2930, 2855, 1785, 1748, 1715, 1370, 1305, 1255, 1154, 1068, 932, 833, 777 cm−1; 1H NMR (400 MHz, CDCl3) δ 4.09 (d, J = 5.2 Hz, 1H), 3.80 (d, J = 5.6 Hz, 1H), 2.65 (dd, J = 17.6, 5.2 Hz, 1H), 2.25 (d, J = 18.0 Hz, 1H), 1.77–1.68 (m, 2H), 1.66–1.57 (m, 3H), 1.56–1.49 (m, 1H), 1.46 (s, 9H), 1.23–1.01 (m, 4H), 0.87–0.81 (m, 1H), 0.79 (s, 9H), 0.01 (s, 3H), 0.00 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3) δ 173.2, 150.4, 82.7, 72.5, 66.6, 42.9, 40.2, 29.8, 28.0, 26.3, 26.2, 25.6, 17.9, −4.5, −4.6 ppm; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C21H39NO4SiNa: 420.2546, found: 420.2549.
(2S,3R)-tert-Butyl 2-benzyl-3-(tert-butyldimethylsilyloxy)-5-oxopyrrolidine-1-carboxylate 4s.
Pale yellow oil (284 mg, 70%). {[α]25D = +10.4 (c 1.00, CHCl3) lit.9j [α]25D = +8.67 (c 1.07, CHCl3); lit.10b [α]23D = +37.9 (c 1.20, CHCl3)}; IR (film): νmax 2959, 2926, 2860, 1787, 1704, 1370, 1312, 1148, 926, 821 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.37–7.31 (m, 2H), 7.30–7.24 (m, 1H), 7.23–7.18 (m, 2H), 4.16 (dd, J = 10.4, 3.2 Hz, 1H), 4.07 (d, J = 5.2 Hz, 1H), 3.17 (dd, J = 13.2, 4.0 Hz, 1H), 2.65 (dd, J = 17.6, 5.2 Hz, 1H), 2.51 (dd, J = 13.2, 10.4 Hz, 1H), 2.31 (d, J = 17.6 Hz, 1H), 1.60 (s, 9H), 0.74 (s, 9H), −0.23 (s, 3H), −0.24 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3) δ 172.8, 150.0, 136.6, 129.3, 128.9, 127.1, 83.0, 69.1, 66.9, 41.4, 38.0, 28.1, 25.5, 17.8, −5.2, −5.3 ppm; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C22H35NO4SiNa: 428.2233, found: 428.2221.
(2S,3R)-tert-Butyl 3-(tert-butyldimethylsilyloxy)-5-oxo-2-(2-phenylethynyl)pyrrolidine-1-carboxylate (2S,3R)-14a.
Pale yellow oil (316 mg, 76%). [α]25D = +19.5 (c 1.50, CHCl3); IR (film): νmax 2959, 2926, 2860, 2205 (very weak), 1790, 1764, 1715, 1468, 1348, 1308, 1255, 1148, 1074, 1019, 915, 838 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.44–7.38 (m, 2H), 7.37–7.30 (m, 3H), 4.81–4.78 (m, 1H), 4.43 (d, J = 5.2 Hz, 1H), 3.01 (dd, J = 17.2, 5.2 Hz, 1H), 2.45 (d, J = 17.2 Hz, 1H), 1.58 (s, 9H), 0.92 (s, 9H), 0.16 (s, 3H), 0.14 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3) δ 171.7, 149.4, 131.7, 128.8, 128.4, 121.9, 85.5, 84.2, 83.4, 70.5, 59.2, 41.9, 28.1, 25.7, 18.0, −4.8 ppm; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C23H34NO4Si: 416.2257, found: 416.2253.
(2S,3R)-tert-Butyl 3-(tert-butyldimethylsilyloxy)-5-oxo-2-(2-m-tolylethynyl)pyrrolidine-1-carboxylate (2S,3R)-14b.
Pale yellow oil (343 mg, 80%). [α]25D = +25.6 (c 1.00, CHCl3); IR (film): νmax 2959, 2920, 2855, 2367 (very weak), 1792, 1766, 1718, 1369, 1348, 1307, 1148, 1079, 1021, 918, 838 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.26–7.15 (m, 4H), 4.81–4.78 (m, 1H), 4.42 (d, J = 4.8 Hz, 1H), 3.02 (dd, J = 17.2, 5.2 Hz, 1H), 2.45 (d, J = 17.2 Hz, 1H), 2.35 (s, 3H), 1.58 (s, 9H), 0.92 (s, 9H), 0.16 (s, 3H), 0.14 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3) δ 171.8, 149.4, 138.1, 132.3, 129.7, 128.8, 128.3, 121.7, 85.7, 83.8, 83.4, 70.5, 59.3, 41.9, 28.1, 25.7, 21.2, 18.0, −4.8 ppm; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C24H36NO4Si: 430.2414, found: 430.2408.
(2S,3R)-tert-Butyl 3-(tert-butyldimethylsilyloxy)-2-(2-(4-fluorophenyl)ethynyl)-5-oxopyrrolidine-1-carboxylate (2S,3R)-14c.
Yellow oil (334 mg, 77%). [α]25D = +18.2 (c 2.00, CHCl3); IR (film): νmax 2959, 2937, 2860, 2227 (very weak), 1791, 1760, 1721, 1600, 1508, 1364, 1307, 1257, 1149, 1090, 1014, 921, 837 cm−1; 19F NMR (376 MHz, CDCl3) δ −109.88 ppm; 1H NMR (400 MHz, CDCl3) δ 7.42–7.36 (m, 2H), 7.06–6.99 (m, 2H), 4.79–4.76 (m, 1H), 4.41 (d, J = 5.2 Hz, 1H), 2.99 (dd, J = 17.2, 5.2 Hz, 1H), 2.44 (d, J = 17.2 Hz, 1H), 1.57 (s, 9H), 0.91 (s, 9H), 0.15 (s, 3H), 0.13 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3) δ 171.7, 162.8 (d, J = 248.9 Hz), 149.4, 133.7, 133.6, 118.0 (d, J = 3.2 Hz), 115.8, 115.6, 84.5, 83.9, 83.5, 70.4, 59.1, 41.9, 28.0, 25.6, 18.0, −4.8 ppm; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C23H33FNO4Si: 434.2163, found: 434.2159.
(2S,3R)-tert-Butyl 3-(tert-butyldimethylsilyloxy)-2-(hept-1-ynyl)-5-oxopyrrolidine-1-carboxylate (2S,3R)-14d.
Pale yellow oil (323 mg, 79%). [α]25D = +26.4 (c 1.00, CHCl3); IR (film): νmax 2957, 2926, 2855, 2090 (very weak), 1793, 1760, 1720, 1472, 1352, 1306, 1256, 1149, 1085, 927, 828 cm−1; 1H NMR (400 MHz, CDCl3) δ 4.56–4.52 (m, 1H), 4.26 (d, J = 5.2 Hz, 1H), 2.92 (dd, J = 17.2, 5.2 Hz, 1H), 2.36 (d, J = 17.2 Hz, 1H), 2.21–2.15 (m, 2H), 1.56 (s, 9H), 1.52–1.45 (m, 2H), 1.39–1.29 (m, 4H), 0.93–0.87 (m, 12H), 0.11 (s, 3H), 0.10 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3) δ 171.9, 149.5, 86.4, 83.1, 75.4, 70.7, 59.0, 41.8, 30.9, 28.1, 28.0, 25.6, 22.1, 18.6, 18.0, 13.9, −4.9 ppm; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C22H40NO4Si: 410.2727, found: 410.2722.
(2S,3R)-tert-Butyl 3-(tert-butyldimethylsilyloxy)-2-(3,3-dimethylbut-1-ynyl)-5-oxopyrrolidine-1-carboxylate (2S,3R)-14e.
Pale yellow oil (277 mg, 70%). [α]25D = +22.7 (c 1.50, CHCl3); IR (film): νmax 2968, 2931, 2866, 2209 (very weak), 1793, 1764, 1723, 1364, 1348, 1306, 1255, 1149, 1085, 938, 821 cm−1; 1H NMR (400 MHz, CDCl3) δ 4.53–4.50 (m, 1H), 4.25–4.22 (m, 1H), 2.90 (dd, J = 16.8, 5.2 Hz, 1H), 2.36 (d, J = 17.2 Hz, 1H), 1.55 (s, 9H), 1.20 (s, 9H), 0.89 (s, 9H), 0.12 (s, 3H), 0.10 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3) δ 172.1, 149.3, 94.4, 83.0, 73.9, 70.7, 59.0, 41.7, 30.8, 28.0, 27.3, 25.6, 18.0, −4.9 ppm; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C21H38NO4Si: 396.2570, found: 396.2560.
(2R,3R)-tert-Butyl 3-(tert-butyldimethylsilyloxy)-5-oxo-2-(2-phenylethynyl)pyrrolidine-1-carboxylate (2R,3R)-14a.
Pale yellow solid (303 mg, 73%), m.p. 133–134 °C. [α]25D = −37.7 (c 1.00, CHCl3); IR (film): νmax 2953, 2926, 2849, 2370 (very weak), 1781, 1698, 1326, 1288, 1255, 1158, 1093, 937, 840 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.45–7.40 (m, 2H), 7.36–7.30 (m, 3H), 5.05 (d, J = 6.8 Hz, 1H), 4.49 (ddd, J = 14.0, 7.2, 6.4 Hz, 1H), 2.80 (dd, J = 16.4, 9.2 Hz, 1H), 2.67 (dd, J = 16.4, 7.2 Hz, 1H), 1.58 (s, 9H), 0.94 (s, 9H), 0.16 (s, 3H), 0.14 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3) δ 170.6, 149.0, 131.7, 128.5, 128.3, 122.6, 86.3, 83.6, 83.3, 66.1, 55.6, 40.8, 28.0, 25.6, 18.0, −4.7, −4.8 ppm; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C23H34NO4Si: 416.2257, found: 416.2257.
(2R,3R)-tert-Butyl 3-(tert-butyldimethylsilyloxy)-5-oxo-2-(2-m-tolylethynyl)pyrrolidine-1-carboxylate (2R,3R)-14b.
Pale yellow oil (322 mg, 75%). [α]25D = −27.2 (c 1.50, CHCl3); IR (film): νmax 2961, 2928, 2855, 2359, 1792, 1721, 1304, 1146, 1107, 932, 827 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.10–7.03 (m, 3H), 7.01–6.97 (m, 1H), 4.88 (d, J = 4.8 Hz, 1H), 4.33 (ddd, J = 9.2, 5.2, 4.4 Hz, 1H), 2.63 (dd, J = 11.2, 6.4 Hz, 1H), 2.51 (dd, J = 11.2, 4.4 Hz, 1H), 2.18 (s, 3H), 1.41 (s, 9H), 0.78 (s, 9H), 0.00 (s, 3H), −0.02 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3) δ 170.5, 149.0, 137.9, 132.3, 129.3, 128.8, 128.2, 122.4, 86.5, 83.5, 82.9, 66.1, 55.6, 40.8, 28.0, 25.7, 21.2, 18.0, −4.7, −4.8 ppm; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C24H36NO4Si: 430.2414, found: 430.2402.
(2R,3R)-tert-Butyl 3-(tert-butyldimethylsilyloxy)-2-(2-(4-fluorophenyl)ethynyl)-5-oxopyrrolidine-1-carboxylate (2R,3R)-14c.
Pale yellow solid (312 mg, 72%), m.p. 105–106 °C. [α]25D = −29.7 (c 1.00, CHCl3); IR (film): νmax 2953, 2930, 2855, 2200 (very weak), 1791, 1721, 1507, 1304, 1255, 1147, 1101, 836 cm−1; 19F NMR (376 MHz, CDCl3) δ −110.55 ppm; 1H NMR (400 MHz, CDCl3) δ 7.43–7.37 (m, 2H), 7.06–6.98 (m, 2H), 5.03 (d, J = 7.2 Hz, 1H), 4.49 (ddd, J = 14.0, 7.6, 6.8 Hz, 1H), 2.78 (dd, J = 16.4, 9.2 Hz, 1H), 2.67 (d, J = 16.4, 7.2 Hz, 1H), 1.57 (s, 9H), 0.93 (s, 9H), 0.15 (s, 3H), 0.14 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3) δ 170.5, 162.6 (d, J = 248.2 Hz), 149.0, 133.6, 133.5, 118.6 (d, J = 3.2 Hz), 115.7, 115.5, 85.2, 83.6, 83.0, 66.0, 55.5, 40.8, 28.0, 25.6, 18.0, −4.7, −4.8 ppm; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C23H33FNO4Si: 434.2163, found: 434.2157.
(2R,3R)-tert-Butyl 3-(tert-butyldimethylsilyloxy)-2-(hept-1-ynyl)-5-oxopyrrolidine-1-carboxylate (2R,3R)-14d.
Pale yellow oil (290 mg, 71%). [α]25D = −25.2 (c 1.00, CHCl3); IR (film): νmax 2957, 2931, 2858, 2245 (very weak), 1793, 1760, 1725, 1332, 1305, 1256, 1142, 904, 838 cm−1; 1H NMR (400 MHz, CDCl3) δ 4.78 (ddd, J = 7.2, 2.4, 1.6 Hz, 1H), 4.36 (ddd, J = 14.0, 7.2, 6.4 Hz, 1H), 2.72 (dd, J = 16.4, 10.0 Hz, 1H), 2.58 (dd, J = 16.4, 7.2 Hz, 1H), 2.24–2.18 (m, 2H), 1.55 (s, 9H), 1.53–1.47 (m, 2H), 1.40–1.31 (m, 4H), 0.94–0.88 (m, 12H), 0.13 (s, 3H), 0.11 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3) δ 170.8, 149.2, 87.0, 83.3, 73.8, 66.0, 55.1, 40.6, 31.0, 28.2, 28.0, 25.6, 22.2, 18.7, 18.0, 13.9, −4.7, −4.8 ppm; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C22H40NO4Si: 410.2727, found: 410.2721.
(2R,3R)-tert-Butyl 3-(tert-butyldimethylsilyloxy)-2-(3,3-dimethylbut-1-ynyl)-5-oxopyrrolidine-1-carboxylate (2R,3R)-14e.
Pale yellow solid (249 mg, 63%), m.p. 120–121 °C. [α]25D = −29.4 (c 0.50, CHCl3); IR (film): νmax 2967, 2931, 2855, 2044 (very weak), 1780, 1697, 1342, 1297, 1255, 1149, 1023, 839 cm−1; 1H NMR (400 MHz, CDCl3) δ 4.78 (d, J = 7.2 Hz, 1H), 4.36 (ddd, J = 14.4, 7.6, 6.8 Hz, 1H), 2.71 (dd, J = 16.4, 10.4 Hz, 1H), 2.57 (dd, J = 16.4, 7.2 Hz, 1H), 1.56 (s, 9H), 1.22 (s, 9H), 0.94 (s, 9H), 0.13 (s, 3H), 0.11 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3) δ 170.9, 149.0, 95.0, 83.2, 72.2, 65.8, 55.1, 40.5, 30.9, 28.0, 27.4, 25.7, 18.0, −4.7, −4.8 ppm; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C21H38NO4Si: 396.2570, found: 396.2565.
(2S,3R)-tert-Butyl 3-(tert-butyldimethylsilyloxy)-5-oxo-2-vinylpyrrolidine-1-carboxylate 17a.
Pale yellow oil (252 mg, 74%). [α]25D = −0.45 (c 2.80, CHCl3); IR (film): νmax 2953, 2926, 2860, 1787, 1715, 1364, 1309, 1255, 1154, 1079, 838 cm−1; 1H NMR (400 MHz, CDCl3) δ 5.82 (ddd, J = 17.2, 10.4, 6.0 Hz, 1H), 5.25–5.17 (m, 2H), 4.47–4.43 (m, 1H), 4.07–4.04 (m, 1H), 2.74 (dd, J = 17.6, 5.2 Hz, 1H), 2.37–2.30 (m, 1H), 1.50 (s, 9H), 0.88 (s, 9H), 0.09 (s, 3H), 0.08 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3) δ 172.6, 149.8, 133.9, 116.6, 82.9, 69.5, 69.4, 41.1, 27.9, 25.6, 18.0, −4.8 ppm; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C17H31NO4SiNa: 364.1920, found: 364.1915.
(2S,3R)-tert-Butyl 2-(but-1-en-2-yl)-3-(tert-butyldimethylsilyloxy)-5-oxopyrrolidine-1-carboxylate 17b.
Pale yellow oil (251 mg, 68%). [α]25D = +2.6 (c 0.50, CHCl3); IR (film): νmax 2957, 2931, 2859, 1790, 1755, 1721, 1474, 1368, 1306, 1257, 1154, 1087, 920, 838 cm−1; 1H NMR (400 MHz, CDCl3) δ 4.90–4.88 (m, 1H), 4.84–4.82 (m, 1H), 4.38–4.34 (m, 1H), 4.04–4.01 (m, 1H), 2.77 (dd, J = 17.6, 5.6 Hz, 1H), 2.30 (d, J = 17.2 Hz, 1H), 2.20–2.00 (m, 2H), 1.48 (s, 9H), 1.16–1.11 (m, 3H), 0.89 (s, 9H), 0.11 (s, 3H), 0.08 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3) δ 173.1, 149.7, 147.3, 108.1, 82.7, 71.5, 68.6, 41.1, 27.9, 26.5, 25.6, 17.9, 12.1, −4.6, −4.7 ppm; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C19H35NO4SiNa: 392.2233, found: 392.2232.
(2S,3S)-tert-Butyl 2-benzyl-3-hydroxy-5-oxopyrrolidine-1-carboxylate 18.
A solution of 4s (240 mg, 0.59 mmol) was stirred in dry THF (3 mL) at room temperature, and then a solution of TBAF (3 mL, 1.0 mol in THF) was added. After stirring for 3 hours, the mixture was quenched with water and extracted with EtOAc for three times. The combined organic layers were washed with brine, dried over anhydrous Na2SO4, filtered and concentrated, the residue was purified by chromatography on a silica gel column to give a crude intermediate (172 mg), which was treated with DMP (502 mg, 1.18 mmol) in dry CH2Cl2 (5 mL) for 30 min. Then the mixture was quenched carefully with a saturated solution of NaHCO3 and Na2S2O3 and separated. The aqueous layer was extracted with DCM three times and the combined organic layers were washed with brine. The organic layer was dried, filtered and concentrated to give crude ketone, which was directly used without further purification. The above crude mixture was dissolved in MeOH (5 mL) and cooled to 0 °C. Then NaBH4 (22 mg, 0.59 mmol) was added in three portions. After stirring for 2 hours from 0 °C–room temperature, the reaction was quenched with aqueous NaHCO3 solution and extracted with DCM three times. The combined organic layers were concentrated and the residue was purified by chromatography on a silica gel column to give 18 (153 mg, 89%) as a white solid. M.p. 122–123 °C; lit.10d m.p. 122–124 °C. {[α]25D = +26.9 (c 1.00, CHCl3), lit.10b [α]24D = +25.1 (c 0.85, CHCl3); lit.10d [α]25D = +25.2 (c 0.86, CHCl3); lit.9j [α]25D = +27.7 (c 1.12, CHCl3)}; IR (film): νmax 3403, 2975, 1770, 1676, 1366, 1287, 1167 cm−1; 1H NMR (400 MHz, CDCl3): δ 7.35–7.22 (m, 5H), 4.56–4.44 (m, 2H), 3.24–3.09 (m, 2H), 2.63 (dd, J = 17.0, 7.2 Hz, 1H), 2.43 (dd, J = 17.0, 8.0 Hz, 1H), 1.51 (s, 9H) ppm; 13C NMR (100 MHz, CDCl3): δ 171.8, 149.8, 137.8, 129.9, 128.6, 126.7, 83.3, 65.6, 62.7, 40.1, 40.0, 28.0 ppm; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C16H21NO4Na: 314.1368, found: 314.1372.
Streptopyrrolidine 5.
A solution of 18 (120 mg, 0.41 mmol) in CF3COOH (1.5 mL) was stirred for 4 h from 0 °C to room temperature. Then the mixture was concentrated and the residue was purified by chromatography on a silica gel column to give 5 (65 mg, 83%) as a white solid. M.p. 133–134 °C. lit.9j m.p. 132–134 °C; lit.20a m.p. 134–135 °C; lit.20b m.p. 133–135 °C {[α]25D = −44.5 (c 0.15, MeOH); lit.9j [α]25D = −44.6 (c 0.05, MeOH); lit.11 [α]25D = −12 (c 0.05, MeOH)}; lit.20a [α]20D = −44 (c 1.0, MeOH)}; lit.20b [α]20D = −43.5 (c 1.0, MeOH)}; IR (film): νmax 3455, 3229, 1693, 1346, 1044 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.38–7.32 (m, 2H), 7.30–7.24 (m, 3H), 5.78 (brs, 1H), 4.51–4.43 (m, 1H), 3.91 (ddd, J = 10.4, 5.6, 4.8 Hz, 1H), 3.07 (dd, J = 13.6, 5.6 Hz, 1H), 2.98 (d, J = 4.8 Hz, 1H), 2.86 (dd, J = 13.6, 8.8 Hz, 1H), 2.68 (dd, J = 17.2, 6.0 Hz, 1H), 2.42 (dd, J = 17.2, 2.4 Hz, 1H) ppm; 13C NMR (100 MHz, CDCl3) δ 175.6, 137.7, 129.0, 128.9, 126.9, 68.7, 60.8, 41.0, 35.4 ppm; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C11H14NO2: 192.1025, found: 192.1010.
(2S,3R)-tert-Butyl 2-(3-(benzyloxy)propyl)-3-(tert-butyldimethylsilyloxy)-5-oxopyrrolidine-1-carboxylate (2S,3R)-19a.
Pale yellow oil (56 mg, 12%). [α]25D = +23.0 (c 0.20, CHCl3); IR (film): νmax 2959, 2924, 2854, 1782, 1717, 1457, 1364, 1309, 1260, 1152, 1076, 1019, 921, 828 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.40–7.31 (m, 5H), 4.55–4.48 (m, 2H), 4.09 (d, J = 5.2 Hz, 1H), 3.96 (dd, J = 9.2, 3.2 Hz, 1H), 3.57–3.47 (m, 2H), 2.78 (dd, J = 17.6, 5.2 Hz, 1H), 2.36 (d, J = 17.6 Hz, 1H), 1.84–1.62 (m, 4H), 1.54 (s, 9H), 0.89 (s, 9H), 0.09 (s, 3H), 0.08 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3) δ 172.6, 150.1, 138.3, 128.4, 127.7, 82.9, 73.1, 69.8, 68.3, 67.9, 28.9, 28.0, 26.4, 25.7, 17.9, −4.6, −4.7 ppm; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C25H41NO5SiNa: 486.2652, found: 486.2653.
(2R,3R)-tert-Butyl 2-(3-(benzyloxy)propyl)-3-(tert-butyldimethylsilyloxy)-5-oxopyrrolidine-1-carboxylate (2R,3R)-19a.
White solid (93 mg, 20%), m.p. 80–82 °C. [α]25D = −38.9 (c 2.42, CHCl3); IR (film): νmax 2959, 2926, 2860, 1787, 1704, 1370, 1312, 1148, 926, 821 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.38–7.27 (m, 5H), 4.55–4.47 (m, 3H), 4.19–4.13 (m, 1H), 3.53–3.46 (m, 2H), 2.65–2.52 (m, 2H), 2.05–1.95 (m, 1H), 1.82–1.64 (m, 3H), 1.53 (s, 9H), 0.91 (s, 9H), 0.10 (s, 3H), 0.09 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3) δ 171.3, 149.8, 138.6, 128.3, 127.5, 127.4, 83.1, 72.8, 70.2, 66.4, 61.4, 40.8, 28.0, 26.7, 25.7, 25.5, 18.0, −4.7, −5.0 ppm; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C25H41NO5SiNa: 486.2652, found: 486.2643.
(2S,3R)-tert-Butyl 2-(4-(benzyloxy)butyl)-3-(tert-butyldimethylsilyloxy)-5-oxopyrrolidine-1-carboxylate (2S,3R)-19d.
Yellow oil (129 mg, 27%). [α]25D = +24.7 (c 1.50, CHCl3); IR (film): νmax 2959, 2930, 2856, 1786, 1753, 1714, 1368, 1310, 1256, 1152, 1102, 1076, 837 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.29–7.19 (m, 5H), 4.44–4.42 (m, 2H), 3.99 (d, J = 3.2 Hz, 1H), 3.85 (dd, J = 5.6, 2.8 Hz, 1H), 3.43–3.39 (m, 2H), 2.67 (dd, J = 12.0, 3.2 Hz, 1H), 2.28 (d, J = 12.0 Hz, 1H), 1.65–1.55 (m, 4H), 1.46 (s, 9H), 1.38–1.32 (m, 2H), 0.80 (s, 9H), 0.01 (s, 3H), 0.00 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3) δ 172.6, 150.1, 138.5, 128.4, 127.6, 82.8, 73.0, 69.8, 68.3, 67.9, 41.7, 31.9, 29.7, 28.1, 25.7, 22.8, 17.9, −4.6, −4.7 ppm; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C26H44NO5Si: 478.2989, found: 478.2983.
(2R,3R)-tert-Butyl 2-(4-(benzyloxy)butyl)-3-(tert-butyldimethylsilyloxy)-5-oxopyrrolidine-1-carboxylate (2R,3R)-19d.
Pale yellow oil (124 mg, 26%). [α]25D = +45.0 (c 0.40, CHCl3); IR (film): νmax 2953, 2930, 2855, 1789, 1755, 1717, 1368, 1305, 1255, 1162, 839 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.21–7.10 (m, 5H), 4.36–4.32 (m, 2H), 4.06 (dd, J = 5.2, 4.0 Hz, 1H), 3.79–3.73 (m, 1H), 3.36–3.28 (m, 2H), 2.15 (ddd, J = 10.0, 5.6, 4.8 Hz, 1H), 1.89–1.84 (m, 1H), 1.55–1.45 (m, 4H), 1.37 (s, 9H), 1.28–1.19 (m, 2H), 0.74 (s, 9H), 0.00 (s, 3H), −0.03 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3) δ 172.8, 150.3, 138.5, 128.4, 127.6, 127.5, 83.0, 73.0, 71.2, 70.1, 55.3, 34.6, 33.0, 29.6, 28.1, 25.7, 21.8, 18.2, −4.5, −5.3 ppm; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C26H44NO5Si: 478.2989, found: 478.2986.
(2R,3R)-tert-Butyl 2-(6-(benzyloxy)hexyl)-3-(tert-butyldimethylsilyloxy)-5-oxopyrrolidine-1-carboxylate (2S,3R)-19e.
Yellow oil (303 mg, 60%). [α]25D = +22.7 (c 2.00, CHCl3); IR (film): νmax 2959, 2930, 2857, 1786, 1748, 1715, 1368, 1310, 1153, 1078, 921, 837 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.39–7.27 (m, 5H), 4.53–4.51 (m, 2H), 4.07 (d, J = 5.2 Hz, 1H), 3.95–3.90 (m, 1H), 3.51–3.46 (m, 2H), 2.78 (dd, J = 17.6, 5.2 Hz, 1H), 2.34 (d, J = 17.6 Hz, 1H), 1.73–1.60 (m, 3H), 1.55 (s, 9H), 1.46–1.34 (m, 7H), 0.89 (s, 9H), 0.09 (s, 3H), 0.08 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3) δ 172.7, 150.1, 138.6, 128.4, 127.6, 127.5, 82.8, 72.9, 70.3, 68.3, 67.9, 41.7, 29.7, 29.4, 28.1, 26.1, 26.0, 25.7, 17.9, −4.6, −4.7 ppm; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C28H48NO5Si: 506.3302, found: 506.3296.
(2S,3R)-tert-Butyl 3-(tert-butyldimethylsilyloxy)-2-(3-hydroxypropyl)-5-oxopyrrolidine-1-carboxylate 20.
To a solution of 4m (4.9 g, 13.26 mmol) in t-BuOH/H2O (60 mL, v/v = 3/1) were added N-methylmorpholine N-oxide (5.4 g, 39.79 mmol) and potassium osmate(VI) dihydrate aqueous (245 mg, 0.66 mmol). After stirring overnight, the reaction mixture was quenched with a saturated aqueous solution of NaHSO4 and stirred for another 1 h. Then the resulted mixture was concentrated and residue was diluted with water. The mixture was extracted with EtOAc (150 mL × 3) and the combined organic extracts were washed with brine. The organic layer was dried, filtered and concentrated to give crude middle compound without further purification, which was dissolved in THF/H2O (180 mL, v/v = 1/1) and cooled to 0 °C. Then sodium periodate (5.7 g, 26.52 mmol) was added in one portion. After stirring for 1.5 h, the reaction was quenched with Na2S2O3. The mixture was extracted with EtOAc (150 mL × 3) and the combined organic extracts were washed with brine. The organic layer was dried, filtered and concentrated and the residue was purified by flash chromatography on a silica gel column (PE/EA = 1/1) to give 20 (4.31 g, 87%) as a colorless oil. [α]25D = +28.2 (c 1.00, CHCl3); IR (film): νmax 2953, 2932, 2849, 1770, 1715, 1364, 1293, 1140, 1074, 833 cm−1; 1H NMR (400 MHz, CDCl3) δ 4.11–4.05 (m, 1H), 4.00–3.90 (m, 1H), 3.73–3.62 (m, 2H), 2.83–2.73 (m, 1H), 2.40–2.15 (m, 2H), 1.83–1.57 (m, 3H), 1.56–1.47 (m, 10H), 0.89–0.82 (m, 9H), 0.11–0.03 (m, 6H) ppm; 13C NMR (100 MHz, CDCl3) δ 172.7, 150.2, 83.0, 68.3, 67.7, 61.9, 41.7, 28.8, 28.4, 28.0, 25.6, 17.9, −4.6, −4.7 ppm; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C18H36NO5Si: 374.2363, found: 374.2357.
(2S,3R)-tert-Butyl 3-(tert-butyldimethylsilyloxy)-2-(3-(tert-butyldimethylsilyloxy)propyl)-5-oxopyrrolidine-1-carboxylate trans-19b.
To a cooled (0 °C) solution of 20 (4.0 g, 10.70 mmol) TBSCl (2.4 g, 16.05 mmol) and DMAP (1.3 g, 10.70 mmol) in DMF (45 mL) was added imidazole (2.2 g, 32.12 mmol) in one portion. After stirring for 24 h, the mixture was quenched with a saturated aqueous solution of NH4Cl. The resulted mixture was separated and the aqueous phase was extracted with EA (60 mL × 4). The combined organic layers were washed with water (30 mL × 2) and brine, dried and concentrated. The residue was purified by flash chromatography on a silica gel column (PE/EA = 10/1) to give trans-19b (4.86 g, 93%) as a colorless oil. [α]25D = +26.1 (c 1.50, CHCl3); IR (film): νmax 2956, 2930, 2849, 1788, 1716, 1474, 1310, 1256, 1153, 1074, 836 cm−1; 1H NMR (400 MHz, CDCl3) δ 4.08 (d, J = 5.2 Hz, 1H), 3.92 (dd, J = 9.6, 4.0 Hz, 1H), 3.68–3.59 (m, 2H), 2.78 (dd, J = 17.6, 5.2 Hz, 1H), 2.35 (d, J = 17.6 Hz, 1H), 1.81–1.71 (m, 1H), 1.62–1.55 (m, 2H), 1.57 (s, 9H), 1.51–1.42 (m, 1H), 0.89 (s, 9H), 0.88 (s, 9H), 0.09 (s, 3H), 0.08 (s, 3H), 0.05 (s, 6H) ppm; 13C NMR (100 MHz, CDCl3) δ 172.6, 150.0, 82.8, 68.4, 67.9, 62.5, 41.7, 29.3, 28.5, 28.0, 25.9, 25.7, 18.3, 17.9, −4.6, −4.7, −5.3 ppm; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C24H50NO5Si2: 488.3228, found: 488.3229.
(2S,3R,5R)-tert-Butyl 5-allyl-3-(tert-butyldimethylsilyloxy)-2-(3-(tert-butyldimethylsilyloxy)propyl)pyrrolidine-1-carboxylate 2,5-cis25.
A solution of 19b (2.2 g, 4.52 mmol) in dry THF (20 mL) was treated with a solution of LiEt3BH (13.5 mL, 13.56 mmol, 1.0 M in THF) at −78 °C. After stirring for 30 min, the reaction was carefully quenched with MeOH (5 mL) and stirred for another 5 min. Then a solution of sodium bicarbonate was added and the mixture was warmed to room temperature. The resulting mixture was extracted with EtOAc (50 mL × 3) and the combined organic layers were washed with brine. The dried organic layer was filtered and concentrated to give a crude mixture without further purification, which was dissolved in dry CH2Cl2 (40 mL) and cooled to −78 °C. Once 2-allyl-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (1.2 mL, 6.49 mmol) was added, a solution of BF3·OEt2 (2.2 mL, 17.31 mmol) was added slowly and the reaction mixture was stirred from −78 °C to −40 °C overnight. The mixture was quenched with a saturated NaHCO3 solution (10 mL) and warmed to room temperature. The organic phase was separated and the aqueous layer was extracted with CH2Cl2 three times. The combined organic layers were washed with brine, dried and concentrated. The residue was purified by flash chromatography on a silica gel column (PE/EA = 25/1) to give 2,5-cis25 (1.53 g) in 69% yield as a colorless oil. [α]25D = +25.2 (c 0.50, CHCl3); IR (film): νmax 2956, 2930, 2858, 1472, 1391, 1257, 1175, 1078 cm−1; 1H NMR (400 MHz, CDCl3, rotamers) δ 5.82–5.68 (m, 1H), 5.11–5.00 (m, 2H), 4.10–4.05 (m, 1H), 3.88–3.82 (m, 0.5H), 3.75–3.52 (m, 3.5H), 3.00–2.90 (m, 0.5 H), 2.75–2.65 (m, 1H), 2.56–2.45 (m, 0.5H), 2.44–2.34 (m, 0.5H), 2.06–1.95 (m, 1H), 1.90–1.70 (m, 2H), 1.62–1.44 (m, 11H), 1.28–1.14 (m, 1H), 0.94–0.88 (m, 18H), 0.10–0.07 (m, 6H), 0.06–0.04 (m, 6H) ppm; 13C NMR (100 MHz, CDCl3, rotamers) δ 154.0 (153.9), 136.4 (136.3), 116.6 (116.5), 79.0, 76.1 (75.2), 67.8, 63.0, 57.6 (57.3), 38.9 (37.6), 35.5 (34.6), 30.2 (30.1), 29.8 (28.0), 28.6, 25.9, 25.8, 18.3 (18.2), 17.9, −4.7, −4.8, −5.3 ppm; 1H NMR (400 MHz, DMSO, 70 °C) δ 5.75–5.62 (m, 1H), 5.00–4.90 (m, 2H), 4.07–4.03 (m, 1H), 3.70–3.60 (m, 1H), 3.56–3.48 (m, 2H), 3.47–3.40 (m, 1H), 2.77–2.55 (m, 1H), 2.36–2.22 (m, 1H), 2.07–1.98 (m, 1H), 1.70–1.58 (m, 2H), 1.43–1.31 (m, 11H), 1.20–1.13 (m, 1H), 0.82 (s, 9H), 0.81 (s, 9H), 0.01 (s, 6H), −0.04 (s, 6H) ppm; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C27H56NO4Si2: 514.3748, found: 514.3742.
(2S,3R,5R,E)-tert-Butyl 3-(tert-butyldimethylsilyloxy)-2-(3-(tert-butyldimethylsilyloxy)propyl)-5-(4-methoxy-4-oxobut-2-enyl)pyrrolidine-1-carboxylate (5R)-26.
To a solution of 25 (794 mg, 1.55 mmol) and methyl acrylate (2.1 mL, 23.18 mmol) in dry DCM (260 mL) was quickly added Grubbs 2nd generation catalyst (110 mg) and heated to reflux for 7 h, then the mixture was concentrated and the crude was purified by flash chromatography on a silica gel column to give 26 (884 mg, 95%, E:Z = 95:5) as a colorless oil. [α]25D = +28.7 (c 1.50, CHCl3); IR (film): νmax 2955, 2930, 2860, 1750, 1704, 1391, 1255, 1172, 1097, 1057, 837 cm−1; 1H NMR (400 MHz, CDCl3, rotamers) δ 6.92–6.85 (m, 1H), 5.90–5.81 (m, 1H), 4.11–4.07 (m, 1H), 3.97–3.89 (m, 0.5H), 3.87–3.80 (m, 0.5H), 3.76–3.72 (m, 3H), 3.71–3.52 (m, 3H), 3.17–3.08 (m, 0.5H), 2.91–2.82 (m, 0.5H), 2.75–2.65 (m, 0.5H), 2.64–2.52 (m, 0.5H), 2.11–2.00 (m, 1H), 1.91–1.81 (m, 0.5H), 1.79–1.68 (m, 1.5H), 1.60–1.42 (m, 11H), 1.25–1.12 (m, 1H), 0.93–0.87 (m, 18H), 0.10–0.07 (m, 6H), 0.06–0.03 (m, 6H) ppm; 13C NMR (100 MHz, CDCl3, rotamers) δ 167.0 (169.9), 154.0 (153.7), 147.0 (146.9), 122.6, 79.5 (79.3), 76.0 (75.1), 67.7, 63.0, 56.7 (56.5), 51.5 (51.4), 37.7 (36.3), 36.1 (35.2), 30.2 (30.1), 29.8 (28.0), 28.5, 25.9, 25.8, 18.3 (18.2), 17.9, −4.7, −4.8, −5.3 ppm; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C29H58NO6Si2: 572.3803, found: 572.3804.
(2S,3R,5R)-tert-Butyl 3-(tert-butyldimethylsilyloxy)-2-(3-(tert-butyldimethylsilyloxy)propyl)-5-(4-methoxy-4-oxobutyl)pyrrolidine-1-carboxylate (5R)-27.
Compound 26 (800 mg, 1.40 mmol) and 10% Pd/C (80 mg) were stirred overnight under a hydrogen atmosphere. Then the mixture was filtered and concentrated. The residue was purified by flash chromatography on a silica gel column (PE/EA = 10/1) to give 27 (739 mg, 92%) as a colorless oil. [α]25D = +23.4 (c 2.00, CHCl3); IR (film): νmax 2953, 2929, 2858, 1742, 1693, 1391, 1255, 1178, 1098, 1060, 835 cm−1; 1H NMR (400 MHz, CDCl3, rotamers) δ 4.10–4.05 (m, 1H), 3.80–3.48 (m, 7H), 2.42–2.22 (m, 2H), 2.12–2.00 (m, 1.5H), 1.89–1.82 (m, 1H), 1.77–1.68 (m, 2H), 1.67–1.43 (m, 13.5H), 1.24–1.10 (m, 1H), 0.92–0.85 (m, 18H), 0.09–0.03 (m, 12H) ppm; 13C NMR (100 MHz, CDCl3, rotamers) δ 174.1 (174.0), 154.0 (153.9), 79.0, 76.1 (75.2), 67.5, 63.0, 57.6, 51.4, 36.0 (35.2), 34.0 (33.9), 33.8 (32.7), 30.2 (30.1), 29.8 (28.0), 28.6, 25.9, 25.7, 22.2, 18.3 (18.2), 17.9, −4.7, −4.9, −5.3 ppm; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C29H60NO6Si2: 574.3959, found: 574.3954.
(2S,3R,5R)-tert-Butyl 3-(tert-butyldimethylsilyloxy)-2-(3-(tert-butyldimethylsilyloxy)propyl)-5-(5-(dimethoxyphosphoryl)-4-oxopentyl)pyrrolidine-1-carboxylate (5R)-28.
A solution of dimethyl methylphosphonate (0.7 mL, 6.1 mmol) in dry THF (50 mL) was treated with a solution of n-BuLi (2.7 mL, 6.1 mmol, 2.4 M in hexane) at −78 °C for 1 h. Then a solution of compound 27 (500 mg, 0.87 mmol) in THF (10 mL) was slowly added, and the reaction was warmed to −50 °C and stirred for 2 h. The resulted mixture was quenched with a saturated aqueous solution of NH4Cl and extracted with EtOAc (50 mL × 3). The combined organic layers were washed with brine, dried and concentrated. The residue was purified by flash chromatography on a silica gel column (PE/EA = 30/1) to give 28 (563 mg, 97%) as a colorless oil. [α]25D = +23.7 (c 1.50, CHCl3); IR (film): νmax 2955, 2932, 2855, 1750, 1709, 1463, 1393, 1255, 1179, 1035, 835 cm−1; 1H NMR (400 MHz, CDCl3, rotamers) δ 4.08–4.03 (m, 1H), 3.79 (s, 3H), 3.77 (s, 3H), 3.74–3.47 (m, 4H), 3.15–3.08 (m, 1H), 3.07–3.02 (m, 1H), 2.72–2.50 (m, 2H), 2.08–1.98 (m, 1.5H), 1.88–1.66 (m, 3.5H), 1.60–1.44 (m, 13H), 1.22–1.11 (m, 1H), 0.92–0.83 (m, 18H), 0.08–0.02 (m, 12H) ppm; 13C NMR (100 MHz, CDCl3, rotamers) δ 201.8 (201.4), 154.0 (153.9), 79.0, 76.1 (75.2), 67.5, 63.0, 57.7 (57.6), 53.0, 52.9, 44.1 (44.0), 42.0 (41.8), 40.7 (40.5), 35.2 (35.9), 32.3 (33.5), 30.2 (30.0), 29.8 (28.0), 28.6, 25.9, 25.7, 20.5, 17.9 (18.3), −4.7, −4.9, −5.3 ppm; 31P NMR (125 MHz, CDCl3, rotamers) δ 22.89 (22.73) ppm; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C31H65NO8PSi2: 666.3986, found: 666.3983.
(2S,3R,5R,E)-tert-Butyl 3-(tert-butyldimethylsilyloxy)-2-(3-(tert-butyldimethylsilyloxy)propyl)-5-(4-oxoundec-5-enyl)pyrrolidine-1-carboxylate 29.
A solution of sodium hydride (36 mg, 0.90 mmol) in dry THF (8 mL) was carefully treated with a solution of compound 28 (543 mg, 0.75 mmol) at −0 °C. After stirring for 1 h from 0 °C to room temperature, hexaldehyde (0.11 mL, 0.90 mmol) was added and the resulted mixture was stirred for another 3 h. Then the reaction was quenched with a saturated aqueous solution of NH4Cl and extracted with EtOAc (20 mL × 3). The combined organic layers were washed with brine, dried and concentrated. The residue was purified by flash chromatography on a silica gel column (PE/EA = 30/1) to give 29 (459 mg, 88%) as a colorless oil. [α]25D = +30.4 (c 0.50, CHCl3); IR (film): νmax 2953, 2928, 2857, 1692, 1458, 1391, 1356, 1175, 1096, 835 cm−1; 1H NMR (400 MHz, CDCl3, rotamers) δ 6.82–6.77 (m, 1H), 6.12–6.03 (m, 1H), 4.10–4.05 (m, 1H), 3.80–3.47 (m, 4H), 2.67–2.45 (m, 2H), 2.25–2.16 (m, 2H), 2.12–1.98 (m, 1.5H), 1.88–1.81 (m, 1H), 1.78–1.68 (m, 2H), 1.63–1.42 (m, 15.5H), 1.37–1.26 (m, 4H), 1.24–1.11 (m, 1H), 0.92–0.86 (m, 21H), 0.08–0.03 (m, 12H) ppm; 13C NMR (100 MHz, CDCl3, rotamers) δ 200.8 (200.5), 154.1 (153.9), 147.4, 130.4, 79.0, 76.1 (75.2), 67.5, 63.0, 57.7, 39.9, 35.1 (35.9), 32.6 (33.9), 32.4, 31.3, 30.2 (30.1), 29.8 (28.0), 28.6, 27.8, 25.9, 25.7, 22.4, 21.4, 18.3, 17.9, 14.0, −4.7, −4.9, −5.3 ppm; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C35H70NO5Si2: 640.4793, found: 640.4790.
(2S,3R,5R,E)-tert-Butyl 3-(tert-butyldimethylsilyloxy)-2-(3-hydroxypropyl)-5-(4-oxoundec-5-enyl)pyrrolidine-1-carboxylate 30.
Compound 29 (393 mg, 0.61 mmol) was dissolved in mixture of DCM/MeOH (4 mL, v:v = 50:50) and cooled to −51 °C. Then camphorsulfonic acid (100 mg, 0.43 mmol) was added in one portion and the reaction was stirred overnight. The mixture was quenched with TEA (0.61 mmol) and warmed to room temperature. The resulted mixture was extracted with DCM (20 mL × 3) and the combined organic layers were washed with brine, dried and concentrated. The residue was purified by flash chromatography on a silica gel column (PE/EA = 2/1) to give 30 (300 mg, 93%) as a colorless oil. [α]25D = +23.7 (c 1.00, CHCl3); IR (film): νmax 3449, 2956, 2929, 2858, 1688, 1670, 1631, 1393, 1255, 1173, 1054, 843 cm−1; 1H NMR (400 MHz, CDCl3, rotamers) δ 6.87–6.78 (m, 1H), 6.12–6.04 (m, 1H), 4.10–4.01 (m, 1H), 3.81–3.60 (m, 4H), 3.57–3.53 (m, 0.33H), 2.95–2.88 (m, 0.67H), 2.67–2.44 (m, 2H), 2.25–2.16 (m, 2H), 2.13–2.00 (m, 1.33H), 1.90–1.82 (m, 1H), 1.80–1.70 (m, 2H), 1.67–1.57 (m, 2H), 1.56–1.44 (m, 13.67H), 1.35–1.28 (m, 4H), 1.25–1.15 (m, 1H), 0.93–0.87 (m, 12H), 0.08–0.04 (m, 6H) ppm; 13C NMR (100 MHz, CDCl3, rotamers) δ 200.4 (200.8), 154.3 (154.0), 147.5, 130.3, 79.4 (79.1), 75.7 (76.0), 66.1 (67.3), 61.4 (62.6), 57.8, 39.8, 35.8 (35.1), 33.8 (32.6), 32.4, 31.3, 29.0 (29.9), 28.5, 28.2 (29.5), 27.8, 25.7, 22.4, 21.4, 17.9, 13.9, −4.8, −4.9 ppm; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C29H56NO5Si: 526.3928, found: 526.3929.
3-epi-Epohelmin A 3-epi-6.
A solution of alcohol 30 (102 mg, 0.19 mmol) and TEA (0.21 mL, 1.52 mmol) in dry CH2Cl2 (5 mL) was cooled to 0 °C. Then MsCl (43 μL, 0.57 mmol) was added and the mixture was stirred for 30 min. The reaction was quenched with a saturated NH4Cl solution (20 mL) and extracted with CH2Cl2 (25 mL × 3). The combined organic layers were washed with brine for two times, dried and concentrated to give a crude product without further purification. The crude product and 2,6-lutidine (0.11 mL, 0.91 mmol) were dissolved in CH2Cl2 (15 mL) and cooled to −78 °C. The reaction was treated with TESOTf (0.2 mL, 0.87 mmol) and the mixture was allowed to warm to room temperature and the mixture was stirred overnight. The mixture was diluted with water and extracted with CH2Cl2 (30 mL × 3) and the combined organic layers were washed with brine. The resulted organic layer was dried and concentrated to give a crude product without further purification, which was stirred overnight in a mixture of MeOH/HCl (3 mL, v:v = 50:50). Then the mixture was concentrated to give a crude salt, which was dissolved in water (10 mL) and treated with potassium carbonate. The resulted mixture was extracted with CHCl3 (15 mL × 5) and the combined organic layers were dried with MgSO4. After being concentrated, the residue was purified by flash chromatography on a silica gel column (DCM/CH3OH = 80/1–10/1) to give 3-epi-epohelmin A 3-epi-6 (28 mg, 49%), as a yellow oil, [α]25D = +7.8 (c 0.80, CHCl3); IR (film): νmax 3386, 2959, 2927, 2858, 1668, 1627, 1571, 1458, 1407, 1381, 1261, 1177, 1105, 1043 cm−1; 1H NMR (400 MHz, CDCl3) δ 6.84 (ddd, J = 10.4, 5.2, 4.4 Hz, 1H), 6.07 (d, J = 10.8 Hz, 1H), 4.10–4.05 (m, 1H), 3.85–3.80 (m, 1H), 3.28–3.22 (m, 1H), 2.91–2.82 (m, 2H), 2.65–2.55 (m, 2H), 2.49–2.44 (m, 1H), 2.24–2.19 (m, 2H), 2.17–2.10 (m, 1H), 1.96–1.91 (m, 2H), 1.88–1.80 (m, 2H), 1.77–1.70 (m, 2H), 1.68–1.62 (m, 2H), 1.50–1.43 (m, 2H), 1.34–1.28 (m, 4H), 0.91–0.88 (m, 3H) ppm; 13C NMR (100 MHz, CDCl3) δ 200.0, 148.0, 130.2, 75.3, 73.2, 66.6, 53.5, 40.9, 39.4, 33.5, 32.4, 31.4, 29.1, 27.8, 24.6, 22.4, 21.3, 13.9 ppm; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C18H32NO2: 294.2433, found: 294.2424.
Acknowledgements
We thank the National Natural Science Foundation of China (21472022, 21272041, 21072034, 20832005), Key Laboratory for Chemical Biology of Fujian Province for financial support. The authors also thank Dr Han-Qing Dong for helpful suggestions and thank Xiao-Yun, Wei for initial exploration.
References
-
(a) B. M. Trost, Science, 1983, 219, 245 CAS;
(b)
I. Ojima, Catalytic Asymmetric Synthesis, Wiley-VCH, Weinheim, 2000 Search PubMed;
(c)
J. Tsuji, Transition Metal Reagents and Catalysts: Innovations in Organic Synthesis, Wiley, Chichester, 2000 Search PubMed;
(d)
J. G. de Vries and G. A. Molander, in Science of Synthesis Workbench Edition: Stereoselective Synthesis, ed. P. A. Evans, Thieme, Stuttgart, 2011, vol. 1–3 Search PubMed. For selected reviews and accounts, see:
(e) P. A. Wender, V. A. Verma, T. J. Paxton and T. H. Pillow, Acc. Chem. Res., 2008, 41, 40 CrossRef CAS PubMed;
(f) T. Newhouse, P. S. Baran and R. W. Hoffmann, Chem. Soc. Rev., 2009, 38, 3010 RSC;
(g) D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624 CrossRef CAS PubMed;
(h) R. A. Sheldon, Chem. Soc. Rev., 2012, 41, 1437 RSC;
(i) K. M. Engle, T.-S. Mei, M. Wasa and J.-Q. Yu, Acc. Chem. Res., 2012, 45, 788 CrossRef CAS PubMed;
(j) B. G. Hashiguchi, S. M. Bischof, M. M. Konnick and R. A. Periana, Acc. Chem. Res., 2012, 45, 885 CrossRef CAS PubMed.
- For selected recent reviews on p-toluenesulfinimines, see:
(a) P. Zhou, B.-C. Chen and F. A. Davis, Tetrahedron, 2004, 60, 8003 CrossRef CAS PubMed;
(b) D. Morton and R. A. Stockman, Tetrahedron, 2006, 62, 8869 CrossRef CAS PubMed.
- For selected reviews on the chemistry of t-BS-imines, see:
(a) J. A. Ellman, T. D. Owens and T. P. Tang, Acc. Chem. Res., 2002, 35, 984 CrossRef CAS PubMed;
(b) G.-Q. Lin, M.-H. Xu, Y.-W. Zhong and X.-W. Sun, Acc. Chem. Res., 2008, 41, 831 CrossRef CAS PubMed;
(c) F. Ferreira, C. Botuha, F. Chemla and A. Pérez-Luna, Chem. Soc. Rev., 2009, 38, 1162 RSC;
(d) M. T. Robak, M. A. Herbage and J. A. Ellman, Chem. Rev., 2010, 110, 3600 CrossRef CAS PubMed. For selected recent examples, see:
(e) D. M. Hodgson, A. Charlton, R. S. Paton and A. L. Thompson, J. Org. Chem., 2013, 78, 1508 CrossRef CAS PubMed;
(f) H.-C. Xu, S. Chowdhury and J. A. Ellman, Nat. Protoc., 2013, 8, 2271 CrossRef CAS PubMed;
(g) A. W. Buesking, V. Bacauanu, I. Cai and J. A. Ellman, J. Org. Chem., 2014, 79, 3671 CrossRef CAS PubMed;
(h) C. Q. Fontenelle, M. Conroy, M. Light, T. Poisson, X. Pannecoucke and B. Linclau, J. Org. Chem., 2014, 79, 4186 CrossRef CAS PubMed.
- For early selected examples, see:
(a) F. A. Davis and W. McCoull, J. Org. Chem., 1999, 64, 3396 CrossRef CAS;
(b) T. P. Tang, S. K. Volkman and J. A. Ellman, J. Org. Chem., 2001, 66, 8772 CrossRef CAS PubMed;
(c) D. D. Staas, K. L. Savage, C. F. Homnick, N. N. Tsou and R. G. Ball, J. Org. Chem., 2002, 67, 8276 CrossRef CAS PubMed;
(d) J. W. Evans and J. A. Ellman, J. Org. Chem., 2003, 68, 9948 CrossRef CAS PubMed;
(e) Y.-W. Zhong, Y.-Z. Dong, K. Fang, K. Izumi, M.-H. Xu and G.-Q. Lin, J. Am. Chem. Soc., 2005, 127, 11956 CrossRef CAS PubMed.
-
(a) R.-C. Liu, J.-H. Wei, B.-G. Wei and G.-Q. Lin, Tetrahedron: Asymmetry, 2008, 19, 2731 CrossRef CAS PubMed;
(b) C. Xarnod, W. Huang, R.-G. Ren, R.-C. Liu and B.-G. Wei, Tetrahedron, 2012, 68, 6688 CrossRef CAS PubMed.
-
(a) X. Sun, W. Zheng and B.-G. Wei, Tetrahedron Lett., 2008, 49, 6195 CrossRef CAS PubMed;
(b) W. Huang, J.-L. Ye, W. Zheng, H.-Q. Dong and B.-G. Wei, J. Org. Chem., 2013, 78, 11229 CrossRef CAS PubMed.
-
(a) C.-M. Si, W. Huang, Z.-T. Du, B.-G. Wei and G.-Q. Lin, Org. Lett., 2014, 16, 4328 CrossRef CAS PubMed;
(b) C.-M. Si, Z.-Y. Mao, H.-Q. Dong, Z.-T. Du, B.-G. Wei and G.-Q. Lin, J. Org. Chem., 2015, 80, 5824 CrossRef CAS PubMed.
- For books on pyridine alkaloids, see:
(a)
G. B. Fodor and B. Colasanti, The Pyridine and Piperidine Alkaloids: Chemistry and Pharmacology, in Alkaloids: Chemical and Biological Perspectives, ed. S. W. Pelletier, Wiley-Interscience, New York, NY, 1985, vol. 3, pp. 1–90 Search PubMed;
(b)
M. J. Schneider, Pyridine and Piperidine Alkaloids: An Update, in Alkaloids: Chemical and Biological Perspectives, ed. S. W. Pelletier, Pergamon, Oxford, UK, 1996, vol. 10, pp. 155–299 Search PubMed.
- For selected reviews, see:
(a) P.-Q. Huang, Synlett, 2006, 1133 CrossRef CAS;
(b) B. L. Stocker, E. M. Dangerfield, A. L. Win-Mason, G. W. Haslett and M. S. M. Timmer, Eur. J. Org. Chem., 2010, 1615 CrossRef CAS PubMed; For selected examples, see:
(c) R. G. Andrew, R. E. Conrow, J. D. Elliott, W. S. Johnson and S. Ramezani, Tetrahedron Lett., 1987, 28, 6535 CrossRef CAS;
(d) S. Ishibuchi, Y. Ikematsu, T. Ishizuka and T. Kunieda, Tetrahedron Lett., 1991, 32, 3523 CrossRef CAS;
(e) P.-Q. Huang, S.-L. Wang, H. Zheng and X.-S. Fei, Tetrahedron Lett., 1997, 38, 271 CrossRef CAS;
(f) P.-Q. Huang, S.-L. Wang, J.-L. Ye, Y.-P. Ruan, Y.-Q. Huang, H. Zheng and J.-X. Gao, Tetrahedron, 1998, 54, 12547 CrossRef CAS;
(g) G. C. G. Pais and M. E. Maier, J. Org. Chem., 1999, 64, 4551 CrossRef CAS;
(h) Y.-A. Kim, S.-M. Oh and S.-Y. Han, Bull. Korean Chem. Soc., 2001, 22, 327 CAS;
(i) B.-Y. He, T.-J. Wu, X.-Y. Yu and P.-Q. Huang, Tetrahedron: Asymmetry, 2003, 14, 2101 CrossRef CAS;
(j) W. Huang, J.-Y. Ma, M. Yuan, L.-F. Xu and B.-G. Wei, Tetrahedron, 2011, 67, 7829 CrossRef CAS PubMed;
(k) X.-G. Wang, A.-E. Wang and P.-Q. Huang, Chin. Chem. Lett., 2014, 25, 193 CrossRef CAS PubMed.
-
(a) H. Takahata, K. Yamazaki, T. Takamatsu, T. Yamazaki and T. Momose, J. Org. Chem., 1990, 55, 3947 CrossRef CAS;
(b) H. Yoda, H. Yamazaki and K. Takabe, Tetrahedron: Asymmetry, 1996, 7, 373 CrossRef CAS;
(c) P.-Q. Huang, J.-L. Ye, Z. Chen, Y.-P. Ruan and J.-X. Gao, Synth. Commun., 1998, 28, 417 CrossRef CAS PubMed;
(d) H.-Q. Dong and G.-Q. Lin, Chin. J. Chem., 1998, 16, 458 CAS;
(e) D. K. Pyun, B. J. Kim, H. J. Jung, J. H. Kim, J. S. Lee, W. K. Lee and C. H. Lee, Chem. Pharm. Bull., 2002, 50, 415 CrossRef CAS;
(f) N. Fukuda, K. Sasaki, T. V. R. S. Sastry, M. Kanai and M. Shibasaki, J. Org. Chem., 2006, 71, 1220 CrossRef CAS PubMed;
(g) J. Nöth, K. J. Frankowski, B. Neuenswander, J. Aubé and O. Reiser, J. Comb. Chem., 2008, 10, 456 CrossRef PubMed;
(h) T. G. Elford, A. Ulaczyk-Lesanko, G. De Pascale, G. D. Wright and D. G. Hall, J. Comb. Chem., 2009, 11, 155 CrossRef CAS PubMed;
(i) S. Cren, P. Schar, P. Renaud and K. Schenk, J. Org. Chem., 2009, 74, 2942 CrossRef CAS PubMed;
(j) J.-D. Yu, W. Ding, G.-Y. Lian, K.-S. Song, D.-W. Zhang, X. Gao and D. Yang, J. Org. Chem., 2010, 75, 3232 CrossRef CAS PubMed.
- H. J. Shin, T. S. Kim, H.-S. Lee, J. Y. Park, I.-K. Choi and H. J. Kwon, Phytochemistry, 2008, 69, 2363 CrossRef CAS PubMed.
- For the original isolation of epohelmin A, see:
(a) Y. Sakano, M. Shibuya, Y. Yamaguchi, R. Masuma, H. Tomoda, S. Omura and Y. Ebizuka, J. Antibiot., 2004, 57, 564 CrossRef CAS. For the revised the structure of epohelmins A and B, see:
(b) B. B. Snider and X. Gao, Org. Lett., 2005, 7, 4419 CrossRef CAS PubMed;
(c) M. Shibuya, B. B. Snider, Y. Sakano, H. Tomoda, S. Omura and Y. Ebizuka, J. Antibiot., 2005, 58, 599 CrossRef CAS PubMed. For the Asymmetric synthesis of epohelmin B, see:
(d) A. Fürstner and A. Korte, Chem. – Asian J., 2008, 3, 310 CrossRef PubMed.
- As a representative example, see the recently synthetic efforts towards trans-4-substituted-γ-amino-β-hydroxy acids:
(a) A. K. Ghosh, W. Liu, Y. Xu and Z. Chen, Angew. Chem., Int. Ed. Engl., 1996, 35, 74 CrossRef CAS PubMed;
(b) M. Catasús, A. Moyano, M. A. Pericàs and A. Riera, Tetrahedron Lett., 1999, 40, 9309 CrossRef;
(c) M. Haddad, C. Botuha and M. Larchevêque, Synlett, 1999, 1118 CrossRef CAS;
(d) M. E. Maier and C. Hermann, Tetrahedron, 2000, 56, 557 CrossRef CAS;
(e) C. Alemany, J. Bach, J. Garcia, M. López and A. B. Rodríguez, Tetrahedron, 2000, 56, 9305 CrossRef CAS;
(f) C. Palomo, M. Oiarbide, J. M. Garcia, A. Gonzalez, R. Pazos, J. M. Odriozola, P. Banuelos, M. Tello and A. Linden, J. Org. Chem., 2004, 69, 4126 CrossRef CAS PubMed;
(g) C.-F. Dai, F. Cheng, H.-C. Xu, Y.-P. Ruan and P.-Q. Huang, J. Comb. Chem., 2007, 9, 386 CrossRef CAS PubMed;
(h) J.-M. Luo, C.-F. Dai, S. Y. Lin and P.-Q. Huang, Chem. – Asian J., 2009, 4, 328 CrossRef CAS PubMed;
(i) A. K. Ghosh, K. Shurrush and S. Kulkarni, J. Org. Chem., 2009, 74, 4508 CrossRef CAS PubMed;
(j) D. Yoo, J. Song, M. S. Kang, E. S. Kang and Y. G. Kim, Tetrahedron: Asymmetry, 2011, 22, 1700 CrossRef CAS PubMed;
(k) A. Bandyopadhyay, A. Malik, M. G. Kumar and H. N. Gopi, Org. Lett., 2014, 16, 294 CrossRef CAS PubMed.
- K. Stratmann, D. L. Burgoyne, R. E. Moore, G. M. L. Patterson and C. D. Smith, J. Org. Chem., 1994, 59, 7219 CrossRef CAS.
- T. F. Molinski, K. A. Reynolds and B. I. Morinaka, J. Nat. Prod., 2012, 75, 425 CrossRef CAS PubMed.
-
(a) S. Yoshifuji, K. Tanaka, T. Kawai and Y. Nitta, Chem. Pharm. Bull., 1986, 34, 3873 CrossRef CAS;
(b) M. S. Valle, P. Retailleau and C. R. D. Correia, Tetrahedron Lett., 2008, 49, 1957 CrossRef CAS PubMed.
- J. A. Marshall, A. Piettre, M. A. Paige and F. Valeriote, J. Org. Chem., 2003, 68, 1780 CrossRef CAS PubMed.
-
(a) D. B. Dess and J. C. Martin, J. Org. Chem., 1983, 48, 4155 CrossRef CAS;
(b) D. B. Dess and J. C. Martin, J. Am. Chem. Soc., 1991, 113, 7277 CrossRef CAS.
-
(a) D. A. Cogan and J. A. Ellman, J. Am. Chem. Soc., 1999, 121, 268 CrossRef CAS;
(b) G. Liu, D. A. Cogan, T. D. Owens, T. P. Tang and J. A. Ellman, J. Org. Chem., 1999, 64, 1278 CrossRef CAS.
-
(a) J. Poncet, P. Jouin, B. Castro, L. Nicolas, M. Boutar and A. Gaudemer, J. Chem. Soc., Perkin Trans. 1, 1990, 611 RSC;
(b) S.-H. Xiang, H.-Q. Yuan and P.-Q. Huang, Tetrahedron: Asymmetry, 2009, 20, 2021 CrossRef CAS PubMed.
-
(a) P.-Q. Huang, Z.-Q. Guo and Y.-P. Ruan, Org. Lett., 2006, 8, 1435 CrossRef CAS PubMed;
(b) X. Zhou, W.-J. Liu, J.-L. Ye and P.-Q. Huang, J. Org. Chem., 2007, 72, 8904 CrossRef CAS PubMed.
- Z.-H. Chen, Z.-M. Chen, Y.-Q. Zhang, Y.-Q. Tu and F.-M. Zhang, J. Org. Chem., 2011, 76, 10173 CrossRef CAS PubMed.
- J. D. White and S. Jana, J. Org. Chem., 2014, 79, 700 CrossRef CAS PubMed.
-
(a) T. Sengoku, Y. Satoh, M. Oshima, M. Takahashi and H. Yoda, Tetrahedron, 2008, 64, 8052 CrossRef CAS PubMed;
(b) C.-M. Si, Z.-Y. Mao, R.-G. Ren, Z.-T. Du and B.-G. Wei, Tetrahedron, 2014, 70, 7936 CrossRef CAS PubMed.
-
(a) R. H. Grubbs and S. Chang, Tetrahedron, 1998, 54, 4413 CrossRef CAS;
(b) S. Chatterjee, S. Ghadigaonkar, P. Sur, A. Sharma and S. Chattopadhyay, J. Org. Chem., 2014, 79, 8067 CrossRef CAS PubMed.
- CCDC 1417164 (compound trans-4a) contains the supplementary crystallographic data for this paper.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1417164. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5qo00250h |
|
| This journal is © the Partner Organisations 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.