
Aza-Knoevenagel-type condensation of secondary amides: direct access to N-monosubstituted β,β-difunctionalized enamines†‡
Pei-Qiang
Huang
*ab,
Wei
Ou
a and
Jian-Liang
Ye
a
aDepartment of Chemistry, Fujian Provincial Key Laboratory of Chemical Biology, iChEM (Collaborative Innovation Center of Chemistry for Energy Materials), College of Chemistry and Chemical Engineering, Xiamen University, Xiamen, Fujian 361005, P. R. China. E-mail: pqhuang@xmu.edu.cn
bThe State Key Laboratory of Elemento-Organic Chemistry, Nankai University, Tianjin 300071, P. R. China
First published on 20th July 2015
Abstract
An efficient approach to N-monosubstituted β,β-difunctionalized enamines, a class of versatile building blocks for the synthesis of bioactive compounds, is reported. The method is based on the triflic anhydride-mediated direct aza-Knoevenagel-type condensation of secondary amides with active methylene compounds. The reaction showed good chemoselectivity and functional group tolerance. A number of title compounds have been synthesized in good to excellent yields in one pot from readily available starting materials.
Introduction
N-Monosubstituted β,β-difunctionalized enamines1 (1 in Scheme 1) are a sub-class of aza-analogs of the Knoevenagel condensation products (2). Possessing multiple functional groups, these molecules exhibit multiple reactivities, including nucleophilicity at both β-carbon and N-atom, and electrophilicity at both β-functional groups and α-carbon. Consequently, these compounds serve as versatile building blocks in the synthesis of heterocycles,2 alkaloids,3 rigid dipeptide mimetics,4 and medicinal agents5 such as antimalarial,5a antimitotic5b and antibacterial agents,5c inhibitors of hepatitis C virus,5d,e potent inhibitors of lymphocyte-specific kinase,5f and cell-permeable inhibitors of protein tyrosine phosphatase 1B.5g Moreover, such structural motifs are also versatile therapeutic pharmacophores.6 In addition, it has been reported that some of them exhibit interesting bioactivities such as fungicidal activity,7a and some have been developed as potent allosteric modulators of GABAA receptors.7b | ||
| Scheme 1 Two main approaches to the functionalized enamines 1. | ||
Two common approaches for the preparation of N-monosubstituted β,β-difunctionalized enamines 1 are shown in Scheme 1.1–12 These compounds are made either from secondary amides (approach 1),3c,e,5a–c,10–12 or by the transamination or the aminolysis of suitable intermediates (7 and 8, approach 2).2c,3a,b,5d–f,7a,8a,d However, such methods rely on stepwise transformations. For example, approach 1 involves the conversion of an amide 3 into an activated form such as lactim ether3c,e,4,104a/4b, lactim thioether10a4c, imidoyl chloride2e,3c,e,5a–c4d, or thioamide/thiolactam11,125. The activated form of the amide is then converted into 1 by Knoevenagel-type condensation or by the Eschenmoser sulfide contraction method.11 Although recently Hussaini and co-workers have reported the improvements to the Eschenmoser sulfide-contraction,12 that remains a stepwise methodology from amides.
One-pot transformation of secondary amides 3 into functionalized enamines of type 1 is rare. In this context, in their total synthesis of azaphenalene alkaloids, Hsung and co-workers achieved the one-pot synthesis of 10a and 10b from secondary lactams 9a and 9b, respectively3c (Scheme 2, eqn (1)). Recently, Long and co-workers developed a highly efficient synthesis of 2-substituted 3-carboxy-4-quinolone derivatives 11, which involved a one-pot formation of enamino ketoester from secondary amides 3A and aryl ketoesters 6B (Scheme 2, eqn (2)). The low yield of the former method is another reason why a new one-pot method is desirable.
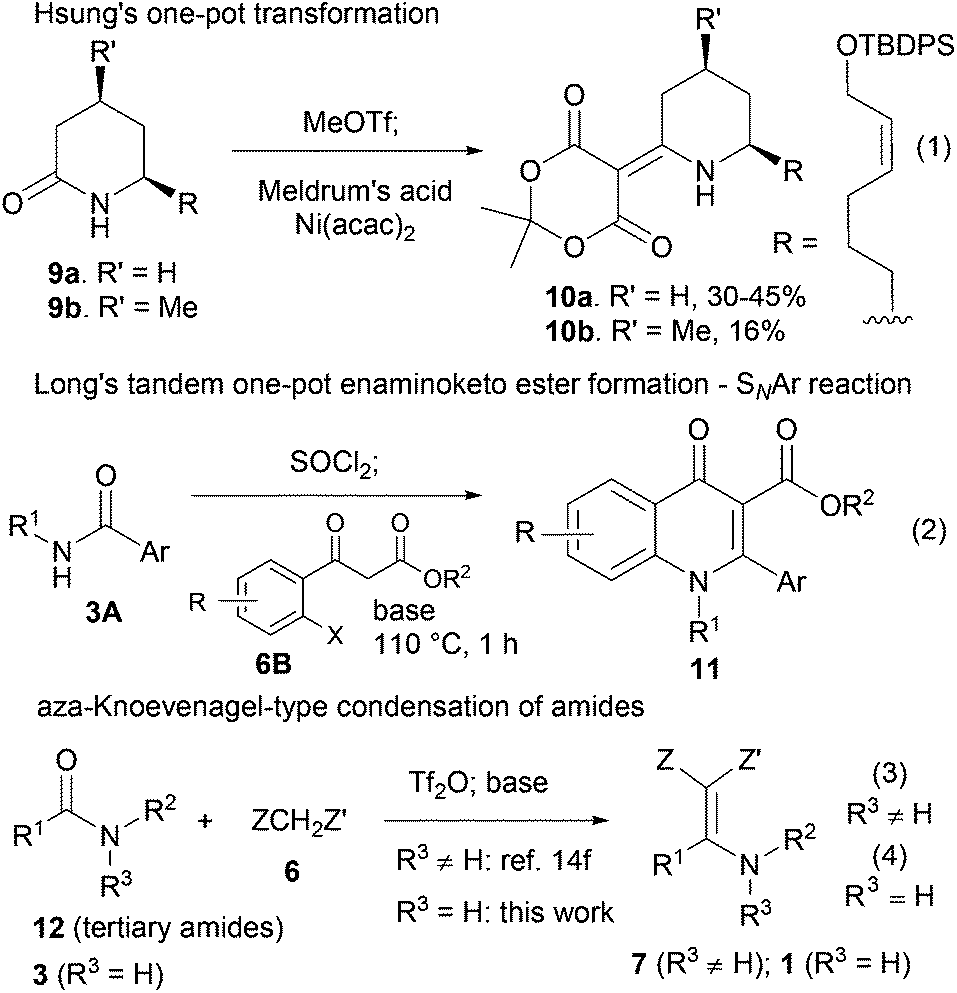 | ||
| Scheme 2 One-pot transformations of secondary amides to functionalized enamines 10, 11 and 1 and our previous approach to 7. | ||
Results and discussion
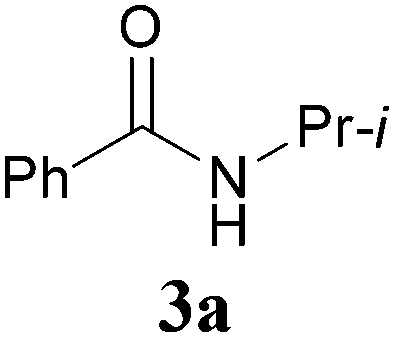
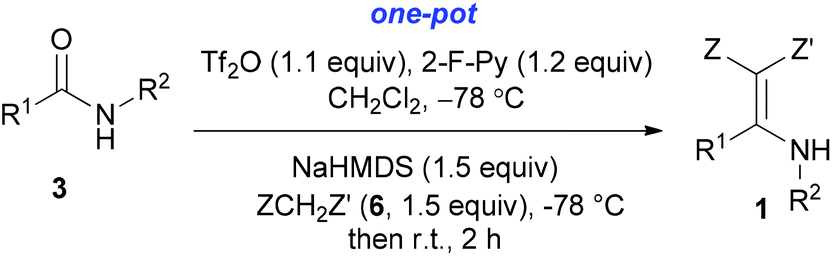
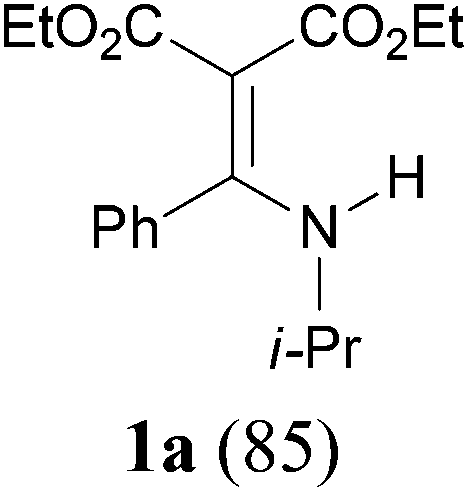
With our longstanding interest in the synthesis of N-containing compounds,13 in recent years, we have focused our attention on the development of synthetic methods based on amide activation.14,15 Within this framework, we discovered very recently a direct Knoevenagel-type condensation reaction of tertiary amides 12 to give N,N-disubstituted β,β-difunctionalized enamines 714f,16 (Scheme 2, eqn (3)). In view of the importance of the Knoevenagel-type condensation reaction products of secondary amides (1), we decided to investigate the Knoevenagel-type condensation reaction of secondary amides (Scheme 2, eqn (4)), and wish to report the results herein.At the outset of our investigation, secondary amide N-isopropyl benzamide 3a was chosen as a model substrate, and the conditions that we established previously for the Knoevenagel-type condensation reaction of tertiary amides14f were adopted. In the event, amide 3a was treated with 1.2 equiv. of triflic anhydride17 (Tf2O) in CH2Cl2 at −78 °C for 30 min and the resulting activated intermediate was subjected to react with carbanion generated in situ from 1.5 equiv. of diethyl malonate (6a) and 2.5 equiv. of sodium hexamethyldisilazane (NaHMDS) at −78 °C, and then at r.t. for 2 h. To our delight, the desired aza-Knoevenagel-type condensation product 1a was obtained in 61% yield (Table 1, entry 1). Decreasing the amount of base from 2.5 equiv. to 1.5 equiv. resulted in a lower yield of 1a (45%) (entry 2). In view of the beneficial effect of 2-fluoropyridine18 (2-F-Py) as a base additive in combination with Tf2O for the activation of secondary amides and subsequent C–C bond forming reaction,14h–j the effect of 2-F-Py on the reaction was examined. Indeed, in the presence of 0.5 and 1.2 equiv. of 2-F-Py, the yields were improved to 63% and 86%, respectively (entries 3 and 4). Lower amounts of NaHMDS were detrimental (entry 5), whereas higher amounts of NaHMDS turned out to be unnecessary (entry 6). Interestingly, sodium hydride and potassium tert-butoxide could also be used as the base, which led to the desired product in 64% and 60% yield, respectively (entries 7 and 8). Thus the optimal conditions for the aza-Knoevenagel-type condensation reaction of secondary amides were determined as: activation of secondary amide (1.0 equiv.) with Tf2O (1.1 equiv.)/2-F-Py (1.2 equiv.) at −78 °C, and the resulting intermediate reacted with diethyl sodiomalonate generated from the corresponding malonate (1.5 equiv.) and NaHMDS (1.5 equiv.) at −78 °C, and then at r.t. for 2 h.
|
|
|||
|---|---|---|---|
| Entry | 2-F-Py (equiv.) | Base (equiv.) | 1a , (% yield) |
| a 1.1 equiv. of Tf2O and 1.5 equiv. of diethyl malonate (6a) were used. b Isolated yield. | |||
| 1 | 0 | NaHMDS (2.5) | 61 |
| 2 | 0 | NaHMDS (1.5) | 45 |
| 3 | 0.5 | NaHMDS (1.5) | 63 |
| 4 | 1.2 | NaHMDS (1.5) | 86 |
| 5 | 1.2 | NaHMDS (1.0) | 80 |
| 6 | 1.2 | NaHMDS (3.0) | 85 |
| 7 | 1.2 | NaH (3.0) | 64 |
| 8 | 1.2 | t-BuOK (3.0) | 60 |

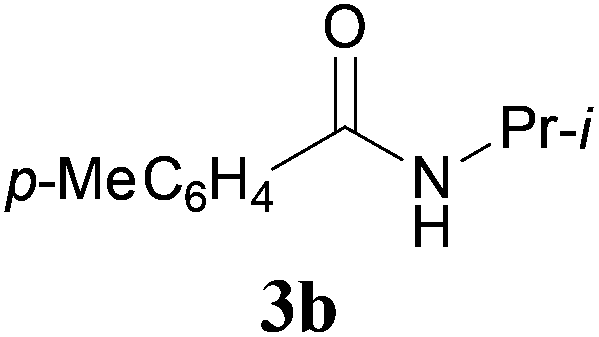
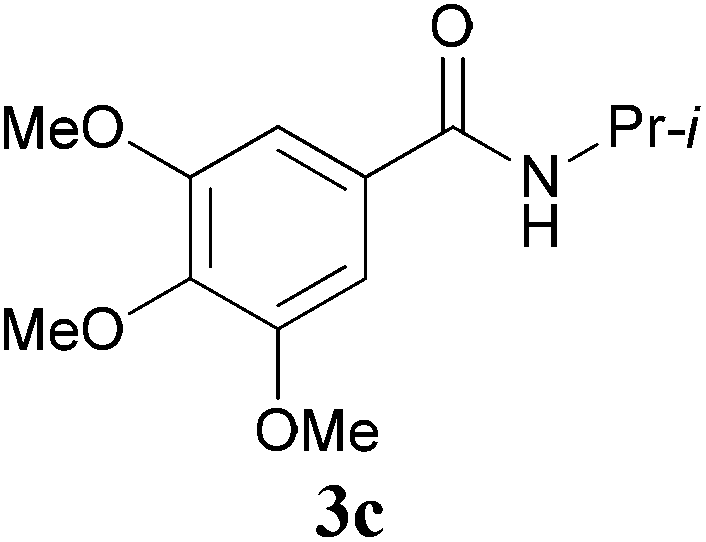
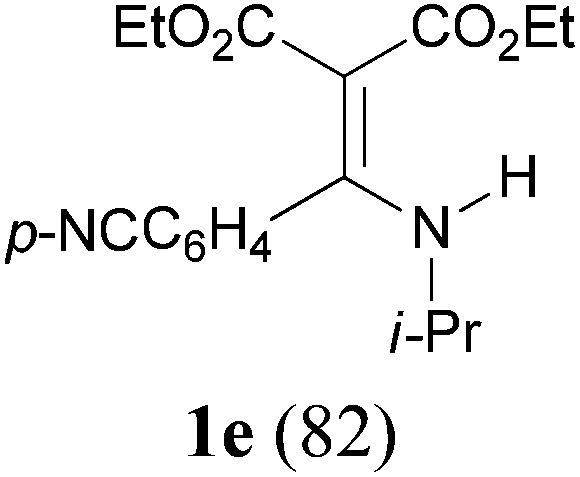
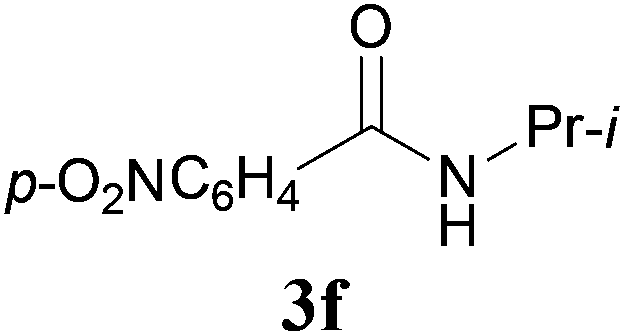
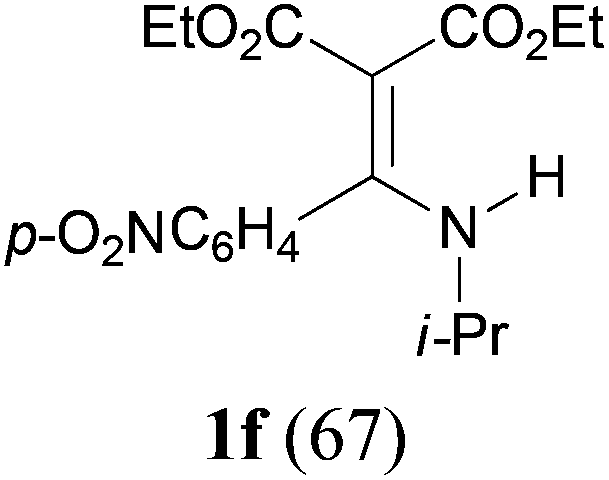
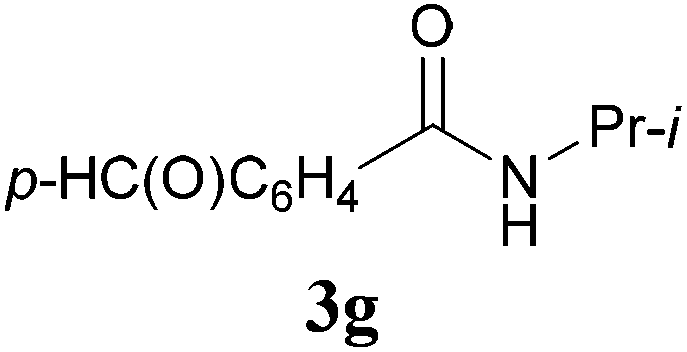
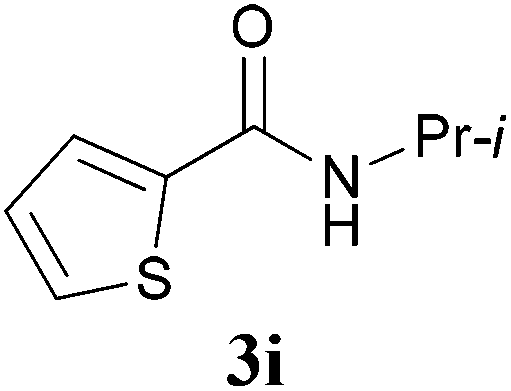
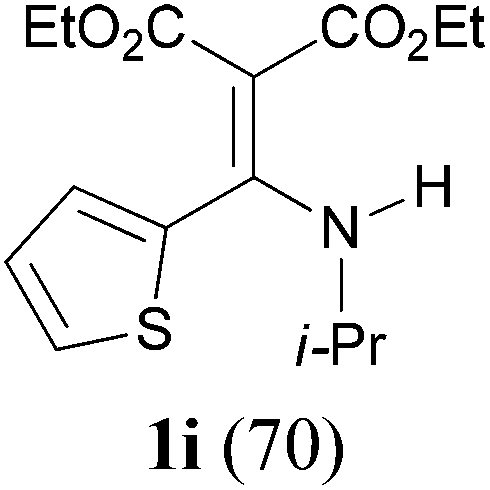
With the optimal conditions in hand, the aza-Knoevenagel-type condensation reactions of different amides were examined, and the results are displayed in Table 2. We first examined the influence of different substituents on the aromatic ring to the outcome of the reaction. The reaction tolerated both electron-donating groups (entries 2 and 3) and electron-withdrawing groups (entries 4–8), and the former are slightly favored over the latter in terms of yield (87% for Me-, 88% for MeO- versus 80% for Cl-substituted benzamide). The reaction of p-nitrobenzamide 3e, which bears a strong electron-withdrawing group (NO2), afforded 1f in a modest yield of 67% (entry 6). Remarkably, as can be seen from entries 5–8, the reaction proceeded chemoselectively at the least reactive secondary amide group in the presence of sensitive functional groups including p-cyano, p-nitro, p-formyl, and p-acetoxy groups to give the corresponding condensation products 1e–1h in 67%–82% yields. Similarly, thiophene-2-carboxamide 3i reacted chemoselectively to produce the corresponding product 1i in 70% (entry 9).



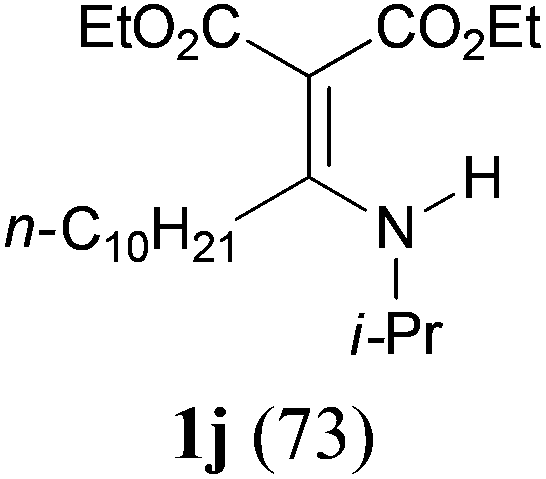
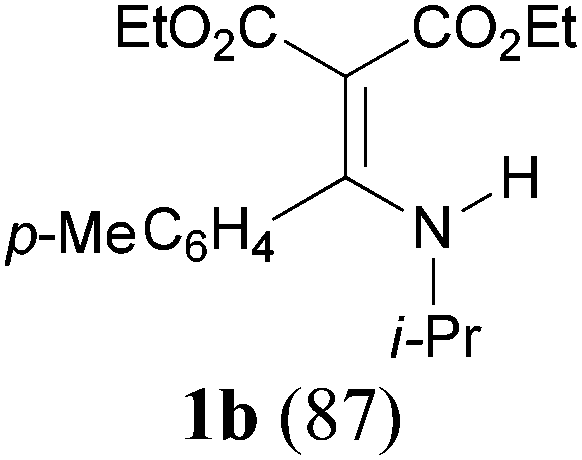
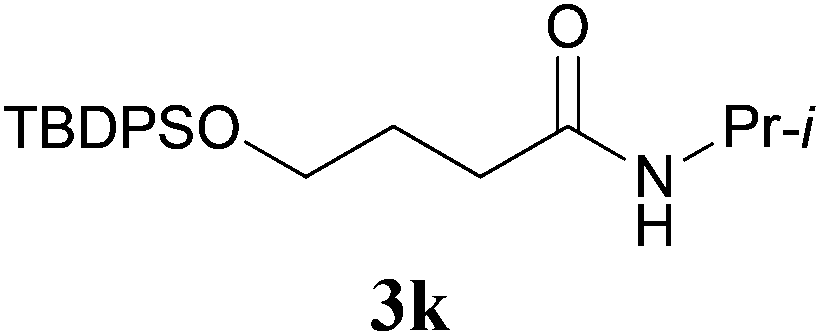

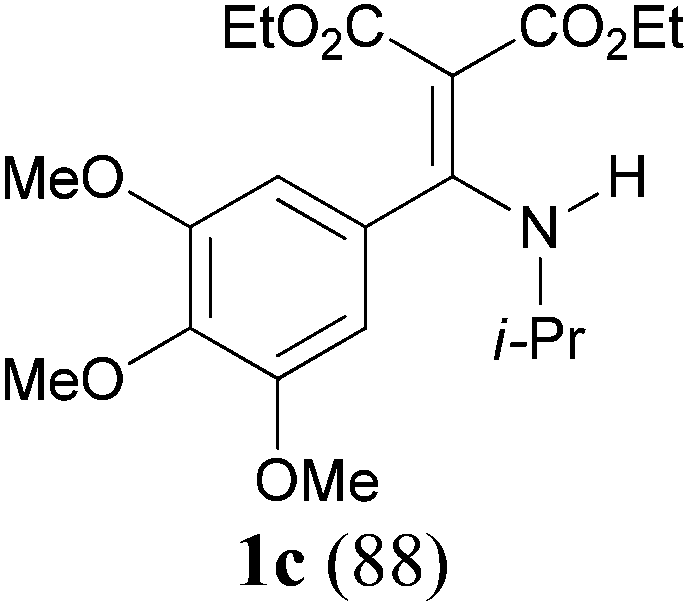
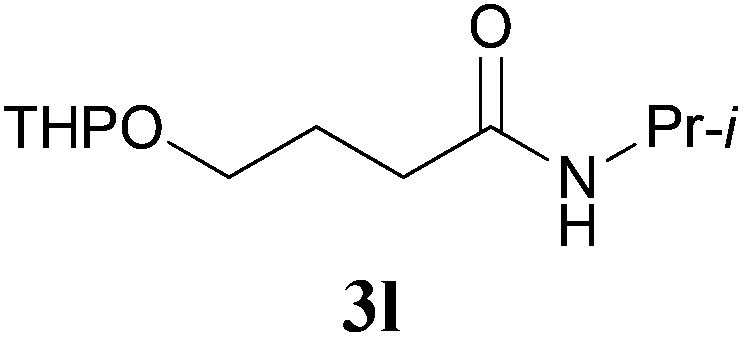






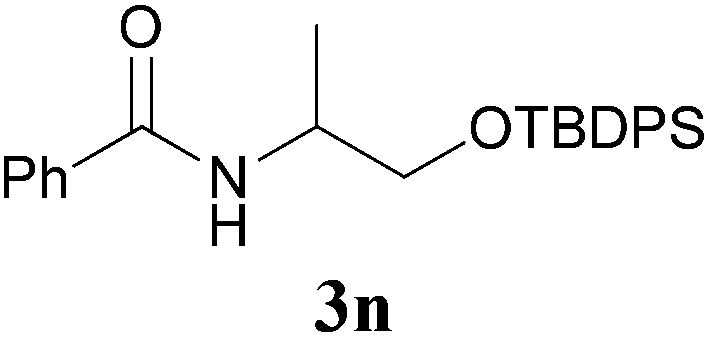

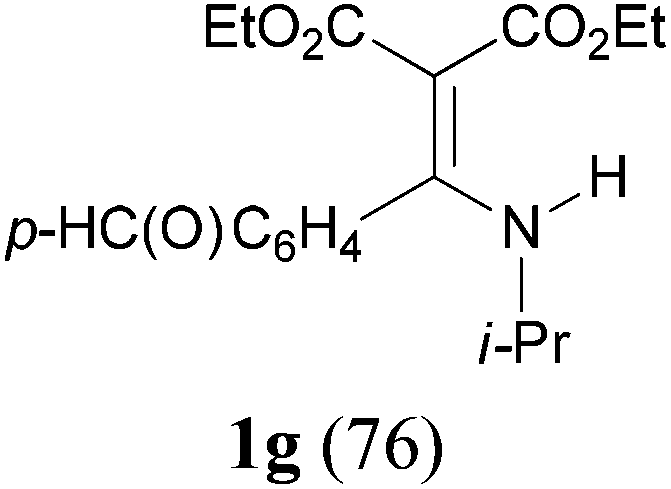


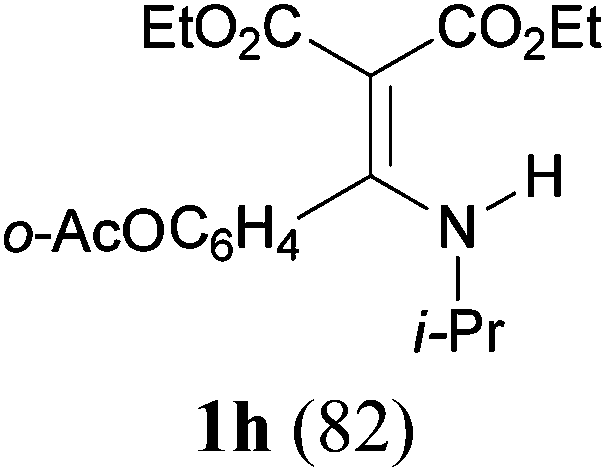


It is worth noting that the reaction is amenable to alkanamides 3j–3o (entries 10–15). Of special importance is the survival of silyl ether (OTBDPS), acetal (OTHP), cyclopropyl, and chloro groups during the condensation reactions of 3k–3o (entries 11–15).
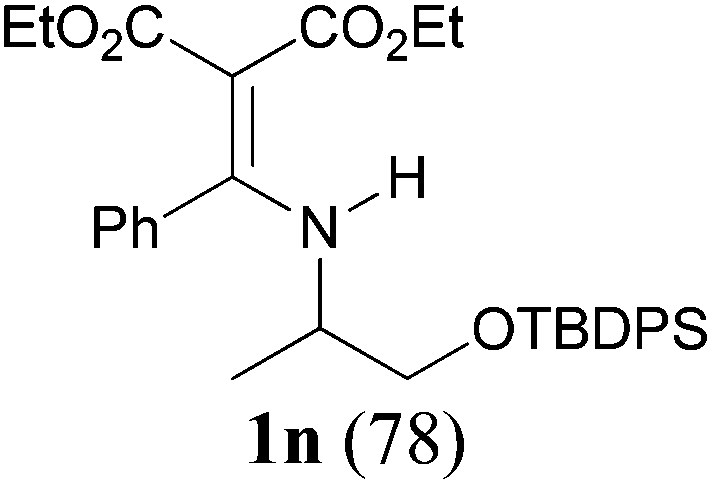
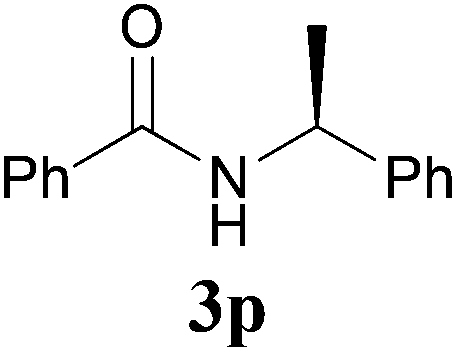
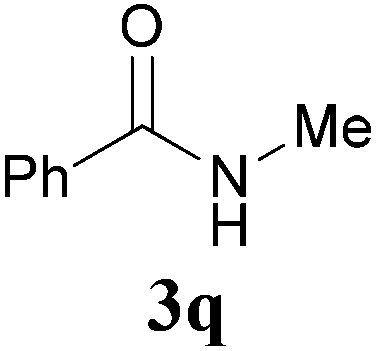
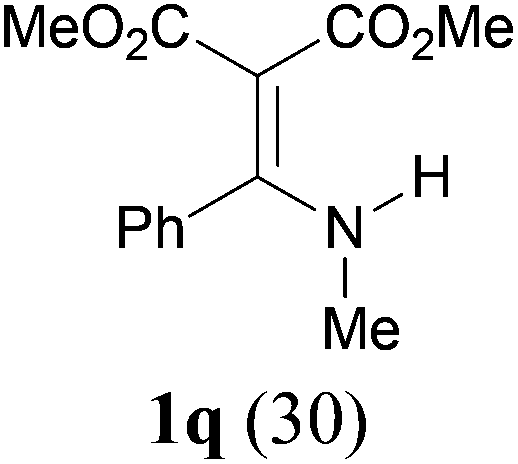
Comparison of the results from the reactions of 3k and 3n showed that a change of the N-alkyl group from the i-propyl to the chiral α-silyloxymethylethyl group did not affect the reaction (entry 11 versus 14, both in 78% yield). The reactions of amides 3o and 3p, derived from (R)- and (S)-α-phenyethylamine, gave the optically active products 1o and 1p in 75% and 80% yield, respectively. However, the reaction of N-methyl benzamide 3q produced the desired product 1q in only 30% yield (entry 17), and the reaction failed with γ-lactam 3r (entry 18).
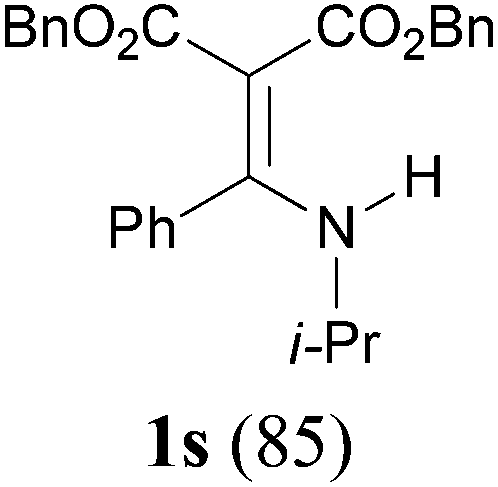
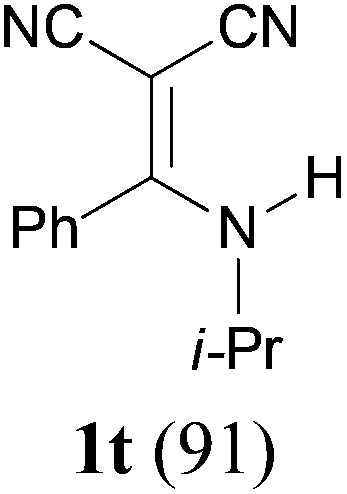
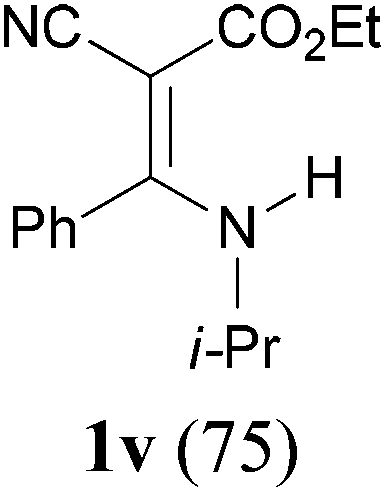
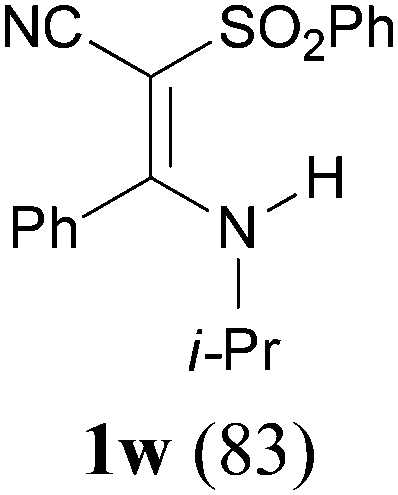
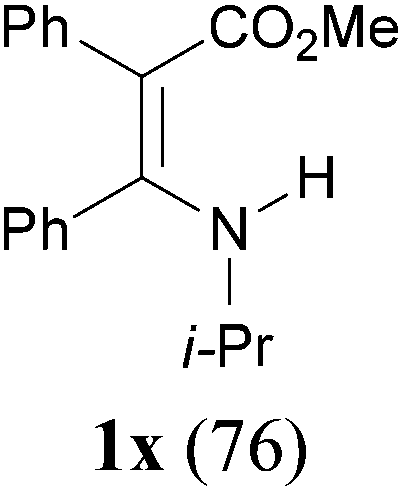
To extend the scope of the reaction, we next investigated the condensation reactions with other active methylene compounds (6) with a methylene group to which two electronegative groups, such as alkoxycarbonyl, phosphonyl, cyano, sulfonyl, and phenyl groups, are attached. More hindered dibenzyl malonate 6b reacted similarly as diethyl malonate 6a did (Table 3, entry 2 versus entry 1). Similarly, the condensation reaction of malononitrile 6c afforded 1t in excellent yield (91%, entry 3). The condensation of ethyl 2-(diethoxyphosphoryl)acetate 6d worked without incidence to afford β-amino vinylphosphonate (β-AVP, 1u)1b,19 as a mixture of two inseparable geometric isomers (E–Z ratio = 62![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :38, determined by integration of the NH in its 1H NMR) in a combined yield of 70% (entry 4). The stereochemistries were not determined. It is worth noting that previously 1u and its related compounds have been synthesized by stepwise methods. These methods were either via transamination or aminolysis reaction between the β-AVPs with a dimethylamine moiety or a β-methoxy vinylphosphonate and a primary amine.1b,19 Interestingly, the condensation reactions of ethyl 2-cyanoacetate 6e and 2-(phenylsulfonyl)acetonitrile 6f produced 1v and 1w, both as a single geometric isomer, in 75% and 83% yield, respectively (entries 5 and 6). The structure of 1v was determined unambiguously by single crystal X-ray diffraction analysis (Fig. 1). It is worth mentioning that the condensation reactions of ethyl 2-phenylacetate 6g and (benzylsulfonyl)benzene 6h, active methylene compounds bearing a weak electron-withdrawing group (phenyl), worked similarly to give the corresponding products 1x and 1y as a single isomer in 76% and 71% yield, respectively (entries 7 and 8). The observed downfield chemical shift of the N–H proton (7.86–9.42 ppm) in the 1H NMR spectra of compounds 1w, 1x and 1y allowed the assignment of the stereochemistries of these compounds as Z-isomers.2c,f,20
:38, determined by integration of the NH in its 1H NMR) in a combined yield of 70% (entry 4). The stereochemistries were not determined. It is worth noting that previously 1u and its related compounds have been synthesized by stepwise methods. These methods were either via transamination or aminolysis reaction between the β-AVPs with a dimethylamine moiety or a β-methoxy vinylphosphonate and a primary amine.1b,19 Interestingly, the condensation reactions of ethyl 2-cyanoacetate 6e and 2-(phenylsulfonyl)acetonitrile 6f produced 1v and 1w, both as a single geometric isomer, in 75% and 83% yield, respectively (entries 5 and 6). The structure of 1v was determined unambiguously by single crystal X-ray diffraction analysis (Fig. 1). It is worth mentioning that the condensation reactions of ethyl 2-phenylacetate 6g and (benzylsulfonyl)benzene 6h, active methylene compounds bearing a weak electron-withdrawing group (phenyl), worked similarly to give the corresponding products 1x and 1y as a single isomer in 76% and 71% yield, respectively (entries 7 and 8). The observed downfield chemical shift of the N–H proton (7.86–9.42 ppm) in the 1H NMR spectra of compounds 1w, 1x and 1y allowed the assignment of the stereochemistries of these compounds as Z-isomers.2c,f,20
 | ||
| Fig. 1 The crystal structure of 1v. | ||


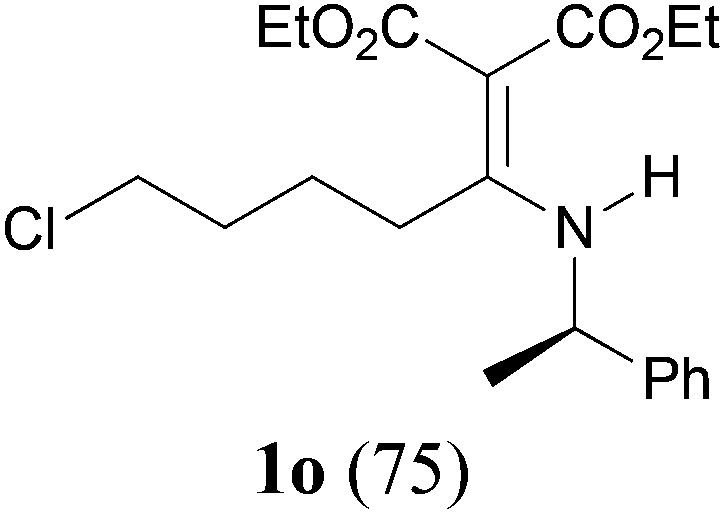
As we have mentioned in the Introduction, the aza-Knoevenagel-type condensation products are multi-functional compounds enabling a number of further reactions. For example, according to the literature precedents,8e–g taking advantage of the nucleophilicity at the nitrogen atom, functionalized secondary chloro enamine 1o (Table 2, entry 15) can be cyclized to give the cyclic tertiary one.21 Moreover, the enantiopure products (R)-1o and (S)-1p (Table 2, entries 15 and 16) are ready precursors for the synthesis of enantiomeric β-amino esters.22
To further demonstrate the utility of the aza-Knoevenagel-type condensation products, the known 1z2c was prepared as a mixture of two inseparable geometric isomers, Z/E = ca. 1:1 (determined by 1H NMR) by the condensation reaction of (R)-N-(1-phenylethyl)acetamide 3s with methyl 2-(2-bromophenyl)-acetate (6i). The conversion of the isomeric 1z into the indole derivative bearing a chiral center (R)-132c can be achieved by Vaswani's Pd-catalyzed C–N ring forming method2c (Scheme 3).
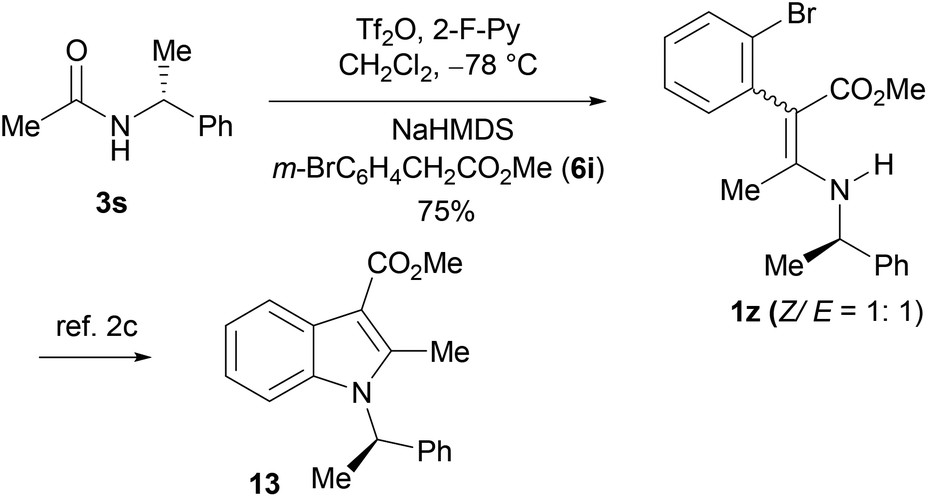 | ||
| Scheme 3 One-pot synthesis of 1z as a ready precursor of functionalized chiral indole (R)-13. | ||
In addition, the condensation reaction of amide 3t with malononitrile (6e) provided 1aa. Then, without further purification, the reaction of crude 1aa with 2-bromoacetophenone in the presence of potassium carbonate provided 14 in 73% yield for two steps (Scheme 4). By using the Takayama's protocol,5f the latter is convertible into poly-substituted pyrrole 15, which belongs to a class of potent inhibitors of lymphocyte-specific kinase.5f
 | ||
| Scheme 4 One-pot synthesis of 1aa as a ready precursor of the lymphocyte-specific kinase inhibitor 15. | ||
Some ethyl Z-2-cyano-3-substituted amino-3-(2-methylphenyl)propenoates such as 1ab have been shown to exhibit fungicidal activities,7a and have been synthesized by a three-step method.7a By our method, the direct condensation of amide 3u with ethyl 2-cyanoacetate 6d produced, in one-pot, 1ab in 82% yield (Scheme 5).
 | ||
| Scheme 5 One-pot synthesis of the fungicidal compound 1ab. | ||
A plausible mechanism of the aza-Knoevenagel-type condensation reaction of secondary amides is depicted in Scheme 6. The nitrogen-lone pair assisted nucleophilic reaction of amide oxygen with triflic anhydride (Tf2O) generates O-triflyl imidate A. Abstraction of the proton in A by a base (NaHMDS or 2-fluoropyridine) generates imidoyl triflate B, which either reacts with sodiomanolate to give directly imine D, or eliminates TfO− to give the nitrilium intermediate C. The fact that secondary γ-lactam 3r failed to undergo the aza-Knoevenagel-type condensation reaction (Table 2, entry 18) precludes the direct substitution with imidoyl triflate B, and thus implicates the intermediacy of the nitrilium intermediate C.
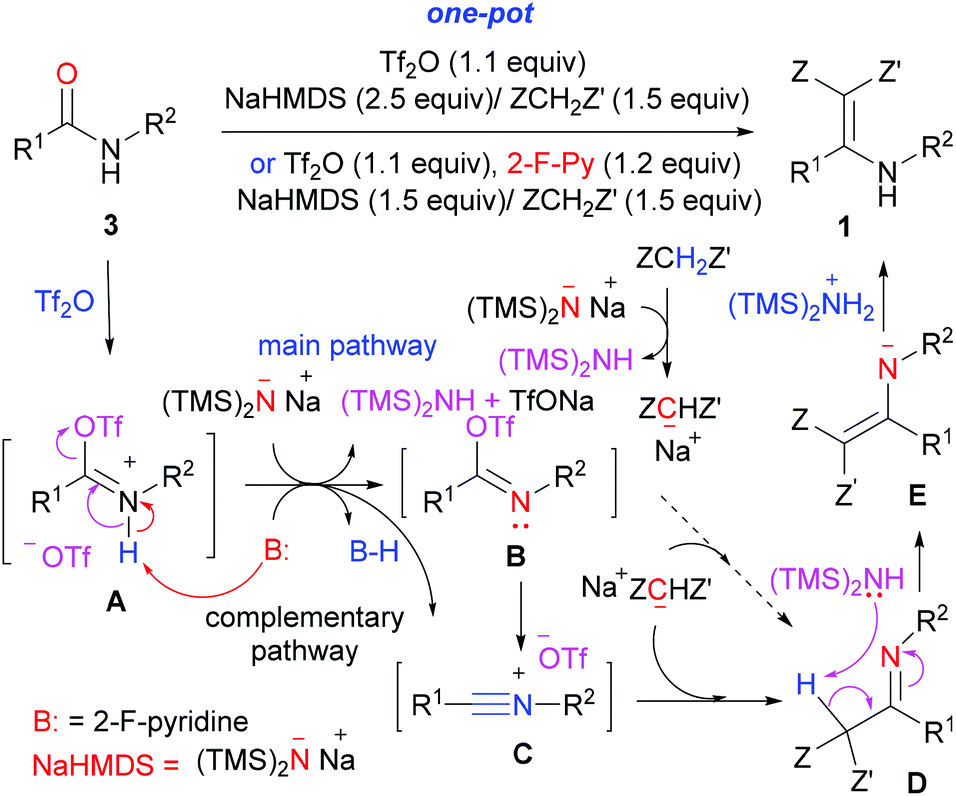 | ||
| Scheme 6 Plausible mechanism of the aza-Knoevenagel-type condensation reaction of secondary amides. | ||
The latter can also be generated directly from O-triflyl imidate A by deprotonation. Nucleophilic addition to nitrilium intermediate C then generates imine D. Finally, a base-promoted step-wise tautomerization of imine D yielded, via enamine N-anion E, the functionalized enamine 1. The key role of 2-fluoropyridine for the generation of the nitrilium intermediate has previously been demonstrated by Movassaghi.18 However, the fact that the reaction proceeds in the absence of 2-fluoropyridine to give the desired product 1 in 61% yield (Table 1, entry 1) implicated that the deprotonation of the intermediate A by NHMDS to generate the nitrilium intermediate C is the main pathway of the reaction.
Conclusion
In summary, we have disclosed a direct, chemoselective, and general synthesis of N-monosubstituted β,β-difunctionalized enamines from readily available secondary amides and active methylene compounds. In conjunction with other efficient synthetic methodologies, such as Vaswani's indole synthesis,2c Zhao's PIFA2f/NXS/Zn(OAc)22d-mediated synthesis of N-substituted indole derivatives, and Takayama's protocol for the synthesis of tetra-substituted pyrroles,5f a number of poly-functionalized medicinally-relevant compounds can be synthesized in a procedure-economical manner.23
Experimental section
General methods
1H NMR and 13C NMR spectra were recorded on a Bruker 400 (1H/400 MHz, 13C/100 MHz) spectrometer. Chemical shifts are expressed in parts per million (δ) relative to an internal standard of residual chloroform (7.26 ppm for 1H NMR and 77.0 ppm for 13C NMR). Data for 1H NMR are reported as chemical shifts (multiplicity, coupling constant, number of proton). ESI-Mass spectra were recorded on a Bruker Dalton ESquire 3000 plus LC-MS apparatus. Optical rotations were measured with a Perkin-Elmer 341 automatic polarimeter or an Anton Paar MCP 500 polarimeter. Melting points were determined on a Büchi M560 Automatic Melting Point apparatus. Infrared spectra were recorded with a Nicolet Avatar 330 FT-IR spectrometer using the film technique. Silica gel (300–400 mesh) was used for flash column chromatography, eluting (unless otherwise stated) with an ethyl acetate/hexane mixture. Tf2O was distilled over phosphorus pentoxide and used within a week. THF was distilled over sodium benzophenone ketyl under N2. Dichloromethane was distilled over calcium hydride under N2.General procedure for the preparation of N-mono-substituted β,β-difunctionalized enamines from secondary amides
Trifluoromethanesulfonic anhydride (185 μL, 1.0 mmol, 1.1 equiv.) was added dropwise to a cooled (−78 °C) solution of an amide (1.0 mmol, 1.0 equiv.) and 2-fluoropyridine (116.5 mg, 103 μL, 1.2 mmol, 1.2 equiv.) in dichloromethane (5 mL). After being stirred for 30 min at −78 °C, a solution of sodium enolate (1.5 mmol, 1.5 equiv.) [freshly prepared by addition of an active methylene compound (1.5 mmol, 1.5 equiv.) to NaHMDS (1.5 mmol, 1.5 equiv.) in tetrahydrofuran (5 mL) and stirred at −78 °C for 1 h] was added dropwise. The resulting mixture was warmed to r.t. and stirred for 2 h at r.t. The reaction was quenched with a saturated aqueous NH4Cl solution and extracted with CH2Cl2 (3 × 10 mL). The combined organic layers were washed with brine, dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel to afford the desired poly-functionalized enamine.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH), 7.10 (dd, J = 1.2, 3.5 Hz, 1H, CH), 7.46 (dd, J = 1.2, 5.0 Hz, 1H, CH), 9.40 (d, J = 9.2 Hz, 1H, NH) ppm; 13C NMR (100 MHz, CDCl3): δ 13.6, 14.3, 24.0 (2C), 46.5, 59.6, 60.3, 95.2, 126.8, 127.6, 128.6, 133.9, 157.3, 167.7, 168.0 ppm; MS (ESI) m/z 334 (M + Na+); HRMS (ESI) m/z calcd for [C15H21NNaO4S]+ (M + Na+): 334.1083; found: 334.1081.
:38, determined by integration of the NH in its 1H NMR; IR (film) νmax: 3195, 2975, 2905, 1739, 1582, 1258, 1030, 959 cm−1. 1H NMR (400 MHz, CDCl3): δ 0.84 (t, J = 7.1 Hz, 3H, CH3), 1.06–1.09 (m, 13H, CH3), 1.33–1.38 (m, 8H, CH3), 3.20–3.28 (m, 1.6H, CH), 3.76 (q, J = 7.1 Hz, 2H, CH2), 3.81–3.86 (m, 1.3H, CH2), 4.14 (q, J = 7.88 Hz, 4H, CH2), 4.14 (q, J = 7.08 Hz, 2.4H, CH2), 7.23–7.42 (m, 8H, Ph–H), 9.72 (d, J = 8.91 Hz, 1H, NH), 10.37 (d, J = 8.82 Hz, 0.6H, NH) ppm; 13C NMR (100 MHz, CDCl3, some peaks overlapped): δ 13.7, 14.2, 16.0 (d, JC–P = 6.7 Hz, 2C), 16.2 (d, J = 6.7 Hz, 2C), 23.4 (2C), 23.7 (2C), 46.2, 46.3, 58.9, 59.9, 60.8 (d, JC–P = 6.1 Hz, 2C), 61.7 (d, JC–P = 5.2 Hz, 2C), 80.9 (d, JC–P = 187.0 Hz, 1C), 83.1 (d, JC–P = 217.1 Hz, 1C), 126.9, 127.8 (d, JC–P = 20.0 Hz, 2C), 128.0, 128.6 (2C), 128.7 (d, JC–P = 30.8 Hz, 2C), 128.9 (2C), 135.0 (d, JC–P = 2.3 Hz, 1C), 136.5 (d, JC–P = 15.2 Hz, 1C), 167.3 (d, JC–P = 11.6 Hz, 1C), 170.6 (d, JC–P = 18.5 Hz, 1C), 170.7 (d, JC–P = 11.4 Hz, 1C), 171.3 (d, JC–P = 10.3 Hz, 1C) ppm; MS (ESI) m/z 392 (M + Na+); HRMS (ESI) m/z calcd for [C18H28NNaO5P]+ (M + Na+): 392.1597; found: 392.1590.
:1 (280 mg, yield: 75%) as a colorless oil; IR (film) νmax: 3250, 2943, 1652, 1591, 1437, 1280, 1245, 1197, 1133, 752, 694 cm−1. 1H NMR (400 MHz, CDCl3): δ 1.50 (s, 3H, CH3), 1.53 (s, 3H, CH3), 1.58 (d, J = 6.8 Hz, 6H, CH3), 3.60 (s, 3H, CH3), 3.61 (s, 3H, CH3), 4.66–4.74 (m, 2H, CH), 7.05–7.12 (m, 3H, Ph–H), 7.18–7.36 (m, 13H, Ph–H), 7.54–7.58 (m, 2H, Ph–H), 9.90 (t, J = 6.9 Hz, 2H) ppm; 13C NMR (100 MHz, CDCl3, some peaks overlapped): δ 16.6, 16.7, 25.10, 25.13, 50.7, 53.2, 53.4, 97.2, 125.4, 125.5, 127.0, 127.1, 127.2, 128.1, 128.4, 128.5, 128.7, 128.8, 132.2, 133.8, 134.0, 139.4, 145.0, 145.1, 169.8, 169.9 ppm; MS (ESI) m/z 374 (M + H+).
CH), 7.10 (dd, J = 1.2, 3.5 Hz, 1H, CH), 7.46 (dd, J = 1.2, 5.0 Hz, 1H, CH), 9.40 (d, J = 9.2 Hz, 1H, NH) ppm; 13C NMR (100 MHz, CDCl3): δ 13.6, 14.3, 24.0 (2C), 46.5, 59.6, 60.3, 95.2, 126.8, 127.6, 128.6, 133.9, 157.3, 167.7, 168.0 ppm; MS (ESI) m/z 334 (M + Na+); HRMS (ESI) m/z calcd for [C15H21NNaO4S]+ (M + Na+): 334.1083; found: 334.1081.
:38, determined by integration of the NH in its 1H NMR; IR (film) νmax: 3195, 2975, 2905, 1739, 1582, 1258, 1030, 959 cm−1. 1H NMR (400 MHz, CDCl3): δ 0.84 (t, J = 7.1 Hz, 3H, CH3), 1.06–1.09 (m, 13H, CH3), 1.33–1.38 (m, 8H, CH3), 3.20–3.28 (m, 1.6H, CH), 3.76 (q, J = 7.1 Hz, 2H, CH2), 3.81–3.86 (m, 1.3H, CH2), 4.14 (q, J = 7.88 Hz, 4H, CH2), 4.14 (q, J = 7.08 Hz, 2.4H, CH2), 7.23–7.42 (m, 8H, Ph–H), 9.72 (d, J = 8.91 Hz, 1H, NH), 10.37 (d, J = 8.82 Hz, 0.6H, NH) ppm; 13C NMR (100 MHz, CDCl3, some peaks overlapped): δ 13.7, 14.2, 16.0 (d, JC–P = 6.7 Hz, 2C), 16.2 (d, J = 6.7 Hz, 2C), 23.4 (2C), 23.7 (2C), 46.2, 46.3, 58.9, 59.9, 60.8 (d, JC–P = 6.1 Hz, 2C), 61.7 (d, JC–P = 5.2 Hz, 2C), 80.9 (d, JC–P = 187.0 Hz, 1C), 83.1 (d, JC–P = 217.1 Hz, 1C), 126.9, 127.8 (d, JC–P = 20.0 Hz, 2C), 128.0, 128.6 (2C), 128.7 (d, JC–P = 30.8 Hz, 2C), 128.9 (2C), 135.0 (d, JC–P = 2.3 Hz, 1C), 136.5 (d, JC–P = 15.2 Hz, 1C), 167.3 (d, JC–P = 11.6 Hz, 1C), 170.6 (d, JC–P = 18.5 Hz, 1C), 170.7 (d, JC–P = 11.4 Hz, 1C), 171.3 (d, JC–P = 10.3 Hz, 1C) ppm; MS (ESI) m/z 392 (M + Na+); HRMS (ESI) m/z calcd for [C18H28NNaO5P]+ (M + Na+): 392.1597; found: 392.1590.
:1 (280 mg, yield: 75%) as a colorless oil; IR (film) νmax: 3250, 2943, 1652, 1591, 1437, 1280, 1245, 1197, 1133, 752, 694 cm−1. 1H NMR (400 MHz, CDCl3): δ 1.50 (s, 3H, CH3), 1.53 (s, 3H, CH3), 1.58 (d, J = 6.8 Hz, 6H, CH3), 3.60 (s, 3H, CH3), 3.61 (s, 3H, CH3), 4.66–4.74 (m, 2H, CH), 7.05–7.12 (m, 3H, Ph–H), 7.18–7.36 (m, 13H, Ph–H), 7.54–7.58 (m, 2H, Ph–H), 9.90 (t, J = 6.9 Hz, 2H) ppm; 13C NMR (100 MHz, CDCl3, some peaks overlapped): δ 16.6, 16.7, 25.10, 25.13, 50.7, 53.2, 53.4, 97.2, 125.4, 125.5, 127.0, 127.1, 127.2, 128.1, 128.4, 128.5, 128.7, 128.8, 132.2, 133.8, 134.0, 139.4, 145.0, 145.1, 169.8, 169.9 ppm; MS (ESI) m/z 374 (M + H+).
Acknowledgements
The authors are grateful for financial support from the NSF of China (21332007 and 21472153), and the Program for Changjiang Scholars and Innovative Research Team in University (PCSIRT) of Ministry of Education.Notes and references
- For related reviews, see: (a) K.-M. Wang, S.-J. Yan and J. Lin, Eur. J. Org. Chem., 2014, 1129 CrossRef CAS PubMed; (b) P. Adler, A. Fadel and N. Rabasso, Tetrahedron, 2014, 70, 4437 CrossRef CAS PubMed; (c) H. M. C. Ferraz and E. R. S. Gonçalo, Quim. Nova, 2007, 30, 957 CrossRef CAS; (d) G. Negri, C. Kascheres and A. J. Kascheres, J. Heterocycl. Chem., 2004, 41, 461 CrossRef CAS PubMed; (e) A.-Z. A. Elassar and A. A. El-Khair, Tetrahedron, 2003, 59, 8463 CrossRef CAS.
- For related reviews, see: (a) Y. Han, J. Sun, Y. Sun, G. Hong and C.-G. Yan, Chin. J. Org. Chem., 2012, 32, 1577 CrossRef CAS; (b) P. Lue and J. V. Greenhill, Heterocycl. Chem., 1996, 67, 207 CAS For selected examples, see: (c) G. Vaswani, B. K. Albrecht, J. E. Audia, A. Côté, L. A. Dakin, M. Duplessis, V. S. Gehling, J.-C. Harmange, M. C. Hewitt, Y. Leblanc, C. G. Nasveschuk and A. M. Taylor, Org. Lett., 2014, 16, 4114 CrossRef PubMed; (d) Q. Yan, J. Luo, D. Zhang-Negrerie, H. Li, X. Qi and K. Zhao, J. Org. Chem., 2011, 76, 8690 CrossRef CAS PubMed; (e) Z. Y. Zheng and H. Alper, Org. Lett., 2008, 10, 4903 CrossRef CAS PubMed; (f) Y. Du, R. Liu, G. Linn and K. Zhao, Org. Lett., 2006, 8, 5919 CrossRef CAS PubMed.
- For selected examples, see: (a) J. D. White, Y. Li and D. C. Ihle, J. Org. Chem., 2010, 75, 3569 CrossRef CAS PubMed; (b) J. D. White and D. C. Ihle, Org. Lett., 2006, 8, 1081 CrossRef CAS PubMed; (c) A. I. Gerasyuto and R. P. Hsung, J. Org. Chem., 2007, 72, 2476 CrossRef CAS PubMed; (d) F. A. Davis, H. Xu and J. Y. Zhang, J. Org. Chem., 2007, 72, 2046 CrossRef CAS PubMed; (e) J. J. Folmer, C. Acero, D. L. Thai and H. Rapoport, J. Org. Chem., 1998, 63, 8170 CrossRef CAS.
- R. Millet, J. Domarkas, P. Rombaux, B. Rigo, R. Houssin and J.-P. Hénichart, Tetrahedron Lett., 2002, 43, 5087 CrossRef CAS.
- (a) Y. Q. Zhang, J. A. Clark, M. C. Connelly, F. Zhu, J. Min, W. A. Guiguemde, A. Pradhan, L. Iyer, A. Furimsky, J. Gow, T. Parman, F. El Mazouni, M. A. Phillips, D. E. Kyle, J. Mirsalis and R. K. Guy, J. Med. Chem., 2012, 55, 4205 CrossRef CAS PubMed; (b) Y.-Y. Lai, L.-J. Huang, K.-H. Lee, Z. Y. Xiao, K. F. Bastow, T. Yamori and S.-C. Kuo, Bioorg. Med. Chem., 2005, 13, 265 CrossRef CAS PubMed; (c) J.-P. Lin and Y.-Q. Long, Chem. Commun., 2013, 49, 5313 RSC; (d) Y.-L. Chen, J. Zacharias, R. Vince, R. J. Geraghty and Z. Q. Wang, Bioorg. Med. Chem., 2012, 20, 4790 CrossRef CAS PubMed; (e) E. Stern, R. Millet, P. Depreux and J.-P. Hénichart, Tetrahedron Lett., 2004, 45, 9257 CrossRef CAS PubMed; (f) T. Takayama, H. Umemiya, H. Amada, T. Yabuuchi, F. Shiozawa, H. Katakai, A. Takaoka, A. Yamaguchi, M. Endo and M. Sato, Bioorg. Med. Chem. Lett., 2010, 20, 108 CrossRef CAS PubMed; (g) Y. Zhi, L.-X. Gao, Y. Jin, C.-L. Tang, J.-Y. Li, J. Li and Y.-Q. Long, Bioorg. Med. Chem., 2014, 22, 3670 CrossRef CAS PubMed.
- (a) N. D. Eddingtona, D. S. Coxa, R. R. Robertsb, J. P. Stablesc, C. B. Powelld and K. R. Scotte, Curr. Med. Chem., 2000, 7, 417 CrossRef; (b) I. O. Edafiogho, S. B. Kombian, K. V. V. Ananthalakshmi, N. N. Salama, N. D. Eddington, T. L. Wilson, M. S. Alexander, P. L. Jackson, C. D. Hanson and K. R. Scott, J. Pharm. Sci., 2007, 96, 2509 CrossRef CAS PubMed.
- (a) L.-G. Wang, F.-Y. Wang, Y.-M. Diao, J.-P. Ni and P. Wei, Chin. J. Org. Chem., 2005, 25, 1254 CAS; (b) D. J. Hogenkamp, T. B. C. Johnstone, J.-C. Huang, W.-Y. Li, M. Tran, E. R. Whittemore, R. E. Bagnera and K. W. Gee, J. Med. Chem., 2007, 50, 3369 CrossRef CAS PubMed.
- For related reviews, see: (a) Y. S. Kudyakova, D. N. Bazhin, M. V. Goryaeva, Y. V. Burgart and V. I. Saloutin, Russ. Chem. Rev., 2014, 83, 120 CrossRef PubMed; (b) H. S. P. Rao, S. Rafi and K. Padmavathy, Tetrahedron, 2008, 64, 8037 CrossRef PubMed; (c) B. Stanovnik and J. Svete, Chem. Rev., 2004, 104, 2433 CrossRef CAS PubMed For selected examples, see: (d) R. Slavik, A. M. Herde, D. Bieri, M. Weber, R. Schibli, S. D. Krämer, S. M. Ametamey and L. J. Mu, Eur. J. Med. Chem., 2014, 80, 554 Search PubMed; (e) J. H. Kim, H. Shin and S.-G. Lee, J. Org. Chem., 2012, 77, 1560 CrossRef CAS PubMed; (f) S. M. Hannick and Y. Kishi, J. Org. Chem., 1983, 48, 3833 CrossRef CAS; (g) J.-P. Célérier, E. Deloisy, P. Kapron, G. Lhommet and P. Maitte, Synthesis, 1981, 130 CrossRef.
- (a) Z.-M. Yan, N. Wu, D. Liang, H.-S. Wang and Y.-M. Pan, Org. Lett., 2014, 16, 4048 CrossRef CAS PubMed; (b) L. Wang, Z. Y. Du, S. Y. Peng, K. Zhang and J. Wang, Adv. Synth. Catal., 2014, 356, 2943 CrossRef CAS PubMed; (c) M. Cordaro, F. Risitano, A. Scala, A. Rescifina, U. Chiacchio and G. Grassi, J. Org. Chem., 2013, 78, 3972 CrossRef CAS PubMed; (d) W. L. Wu, J. Xu, S. J. Huang and W. P. Su, Chem. Commun., 2011, 47, 9660 RSC; (e) T. Takuwa, T. Minowa, J. Y. Onishi and T. Mukaiyama, Bull. Chem. Soc. Jpn., 2004, 77, 1717 CrossRef CAS.
- (a) J. P. Célérier, M. G. Richaud and G. Lhommet, Synthesis, 1983, 195 CrossRef; (b) P. C. Jean, D. Elisabeth, L. Gerard and M. Pierre, J. Org. Chem., 1979, 44, 3089 CrossRef; (c) T. Oishi, M. Nagai, T. Onuma, H. Moriyama, K. Tsutae, M. Ochia and Y. Ban, Chem. Pharm. Bull., 1969, 17, 2306 CrossRef CAS; (d) Z.-I. Horii, K. Morikawa and I. Ninomiya, Chem. Pharm. Bull., 1969, 17, 2230 CrossRef CAS.
- (a) M. Roth, P. Dubs, E. Gotschi and A. Eschenmoser, Helv. Chim. Acta, 1971, 54, 710 CrossRef CAS PubMed. For reviews, see: (b) K. Shiosaki, in Comprehensive Organic Synthesis, ed. B. M. Trost, Pergamon Press, Oxford, 2nd edn, 1991, vol. 2, pp. 865–892 Search PubMed; (c) S. Breveman and M. Cherkinsky, in Comprehensive Organic Synthesis II, ed. P. Knochel and G. A. Molander, Elsevier, Amsterdam, 2014, ch. 3.18, p. 887 Search PubMed; (d) S. R. Hussaini, R. R. Chamala and Z. Wang, Tetrahedron, 2015, 71, 6017 CrossRef PubMed.
- For recent work on the Eschenmoser sulfide contraction, see: (a) N. D. Koduri, B. Hileman, J. D. Cox, H. Scott, P. Hoang, A. Robbins, K. Bowers, L. Tsebaot, K. Miao, M. Castaneda, M. Coffin, G. Wei, T. D. W. Claridge, K. P. Roberts and S. R. Hussaini, RSC Adv., 2013, 3, 181 RSC; (b) N. D. Koduri, Z. G. Wang, G. Cannell, K. Cooley, T. M. Lemma, K. Miao, M. Nguyen, B. Frohock, M. Castaneda, H. Scott, D. Albinescu and S. R. Hussaini, J. Org. Chem., 2014, 79, 7405 CrossRef CAS PubMed; (c) N. D. Koduri, H. Scott, B. Hileman, J. D. Cox, M. Coffin, L. Glicksberg and S. R. Hussaini, Org. Lett., 2012, 14, 440 CrossRef CAS PubMed; (d) B. A. D. Neto, A. A. M. Lapis, A. B. Bernd and D. Russowsky, Tetrahedron, 2009, 65, 2484 CrossRef CAS PubMed.
- For a recent account, see: (a) A.-E. Wang and P.-Q. Huang, Pure Appl. Chem., 2014, 86, 1227 CAS. For recent examples, see: (b) J. Zhang, H.-K. Zhang and P.-Q. Huang, Beilstein J. Org. Chem., 2013, 9, 2358 CrossRef PubMed; (c) X.-G. Wang, A. E. Wang and P.-Q. Huang, Chin. Chem. Lett., 2014, 23, 193 CrossRef PubMed; (d) Z.-Y. Mao, S.-Y. Huang, L.-H. Gao, A.-E Wang and P.-Q. Huang, Sci. China: Chem., 2014, 57, 252 CrossRef CAS; (e) S.-P. Luo, Q.-L. Peng, C.-P. Xu, A.-E Wang and P.-Q. Huang, Chin. J. Chem., 2014, 32, 757 CrossRef CAS PubMed; (f) J.-L. Ye, Y.-F. Zhang, Y. Liu, J.-Y. Zhang, Y.-P. Ruan and P.-Q. Huang, Org. Chem. Front., 2015, 2, 697 RSC , and references cited therein.
- For selected recent examples, see: (a) S.-Y. Huang, Z. Chang, S.-C. Tuo, L.-H. Gao, A.-E Wang and P.-Q. Huang, Chem. Commun., 2013, 49, 7088 RSC; (b) K.-J. Xiao, Y. Wang, Y.-H. Huang, X.-G. Wang and P.-Q. Huang, J. Org. Chem., 2013, 78, 8305 CrossRef CAS PubMed; (c) K.-J. Xiao, J.-M. Luo, X.-E. Xia, Y. Wang and P.-Q. Huang, Chem. – Eur. J., 2013, 19, 13075 CrossRef CAS PubMed; (d) H.-Q. Deng, X.-Y. Qian, Y.-X. Li, J.-F. Zheng, L. Xie and P.-Q. Huang, Org. Chem. Front., 2014, 1, 258 RSC; (e) L.-H. Gao, A.-E Wang and P.-Q. Huang, Sci. China: Chem., 2014, 57, 252 CrossRef; (f) P.-Q. Huang, W. Ou, K.-J. Xiao and A.-E Wang, Chem. Commun., 2014, 50, 8761 RSC; (g) P.-Q. Huang and H. Geng, Org. Chem. Front., 2015, 2, 150 RSC; (h) P.-Q. Huang, Q.-W. Lang, A.-E Wang and J.-F. Zheng, Chem. Commun., 2015, 51, 1096 RSC; (i) P.-Q. Huang, Y.-H. Huang, K.-J. Xiao, Y. Wang and X.-E. Xia, J. Org. Chem., 2015, 80, 2861 CrossRef CAS PubMed; (j) J.-F. Zheng, X.-Y. Qian and P.-Q. Huang, Org. Chem. Front., 2015, 2, 927 RSC , and references cited therein.
- For recent reviews and perspectives, see: (a) V. Pace, W. Holzer and B. Olofsson, Adv. Synth. Catal., 2014, 356, 3697 CrossRef CAS PubMed; (b) T. Sato and N. Chida, Org. Biomol. Chem., 2014, 12, 3147 RSC; (c) V. Pace and W. Holzer, Aust. J. Chem., 2013, 66, 507 CAS; (d) C. Madelaine, V. Valerio and N. Maulide, Chem. – Asian J., 2011, 6, 2224 CrossRef CAS PubMed; (e) D. Seebach, Angew. Chem., Int. Ed., 2011, 50, 96 CrossRef CAS PubMed. For selected recent methods, see: (f) P. Jakubec, A. Hawkins, W. Felzmann and D. J. Dixon, J. Am. Chem. Soc., 2012, 134, 17482 CrossRef CAS PubMed; (g) Y. Yanagita, H. Nakamura, K. Shirokane, Y. Kurosaki, T. Sato and N. Chida, Chem. – Eur. J., 2013, 19, 678 CrossRef CAS PubMed; (h) G. Vincent, D. Karila, G. Khalil, P. Sancibrao, D. Gori and C. Kouklovsky, Chem. – Eur. J., 2013, 19, 9358 CrossRef CAS PubMed; (i) K. Shirokane, T. Wada, M. Yoritate, R. Minamikawa, N. Takayama, T. Sato and N. Chida, Angew. Chem., Int. Ed., 2014, 53, 512 CrossRef CAS PubMed; (j) M. Nakajima, Y. Oda, T. Wada, R. Minamikawa, K. Shirokane, T. Sato and N. Chida, Chem. – Eur. J., 2014, 20, 17565 CrossRef CAS PubMed; (k) M. Nakajima, T. Sato and N. Chida, Org. Lett., 2015, 17, 1696 CrossRef CAS PubMed; (l) A. W. Gregory, A. Chambers, A. Hawkins, P. Jakubec and D. J. Dixon, Chem. – Eur. J., 2015, 21, 111 CrossRef CAS PubMed; (m) K. Shirokane, Y. Tanaka, M. Yoritate, N. Takayama, T. Sato and N. Chida, Bull. Chem. Soc. Jpn., 2015, 88, 522 CrossRef CAS , and references cited therein.
- For previous works related to ref. 14f, see: (a) G. Bélanger, R. Larouche-Gauthier, F. Ménard, M. Nantel and F. Barabé, J. Org. Chem., 2006, 71, 704 CrossRef PubMed; (b) G. Bélanger, R. Larouche-Gauthier, F. Ménard, M. Nantel and F. Barabé, Org. Lett., 2005, 7, 4431 CrossRef PubMed; (c) A. Jackson and O. Meth-Cohn, J. Chem. Soc., Chem. Commun., 1995, 1319 RSC; (d) A. G. Martinez, R. M. Alvarez, J. O. Barcina, S. M. Cerero, E. T. Vilar, A. G. Fraile, M. Hanack and L. R. Subramanian, J. Chem. Soc., Chem. Commun., 1990, 1571 RSC.
- For a recent review on the chemistry of Tf2O, see: (a) I. L. Baraznenok, V. G. Nenajdenko and E. S. Balenkova, Tetrahedron, 2000, 56, 3077 CrossRef CAS. For recent examples on the use of Tf2O as a powerful amide activator, see: (b) J. W. Medley and M. Movassaghi, Angew. Chem., Int. Ed., 2012, 51, 4572 CrossRef CAS PubMed; (c) J. W. Medley and M. Movassaghi, Org. Lett., 2013, 15, 3614 CrossRef CAS PubMed; (d) V. Valerio, D. Petkova, C. Madelaine and N. Maulide, Chem. – Eur. J., 2013, 19, 2606 CrossRef CAS PubMed; (e) B. Peng, D. Geerdink and N. Maulide, J. Am. Chem. Soc., 2013, 135, 14968 CrossRef CAS PubMed; (f) B. Peng, D. Geerdink, C. Farès and N. Maulide, Angew. Chem., Int. Ed., 2014, 53, 5462 CrossRef CAS PubMed; (g) W. S. Bechara, I. S. Khazhieva, E. Rodriguez and A. B. Charette, Org. Lett., 2015, 17, 1184 CrossRef CAS PubMed , and references cited therein.
- J. W. Medley and M. Movassaghi, J. Org. Chem., 2009, 74, 1341 CrossRef CAS PubMed.
- (a) M. Pięta, J. Kędzia and T. Janecki, Tetrahedron Lett., 2015, 56, 1891 CrossRef PubMed; (b) J. Kędzia, J. Modranka and T. Janecki, Tetrahedron Lett., 2011, 52, 6623 CrossRef PubMed; (c) T. Janecki, A. Albrecht, J. F. Koszuk, J. Modranka and D. Slowak, Tetrahedron Lett., 2010, 51, 2274 CrossRef CAS PubMed; (d) E. E. Aboujaoude, N. Collignon and P. Savignac, Tetrahedron, 1985, 41, 427 CrossRef CAS.
- (a) T. Hayashi, I. Hori, H. Baba and H. Midorikawa, Bull. Chem. Soc. Jpn., 1967, 40, 2160 CrossRef CAS; (b) L. Casarrubios, J. A. Pérez, M. Brookhart and J. L. Templeton, J. Org. Chem., 1996, 61, 8358 CrossRef CAS; (c) A. F. Shidlovskii, A. S. Peregudov, B. B. Averkiev, M. Y. Antipin and N. D. Chkanikov, Russ. Chem. Bull. Int. Ed., 2004, 53, 2060 CrossRef CAS.
- (a) T. Ponpandian and S. Muthusubramanian, Tetrahedron, 2013, 69, 527 CrossRef CAS PubMed; (b) S. Calvet-Vitale, C. Vanucci-Bacqué, M.-C. Fargeau-Bellassoued and G. Lhommet, J. Org. Chem., 2006, 71, 2071 CrossRef CAS PubMed; (c) M.-X. Zhao, M.-X. Wang, C.-Y. Yu, Z.-T. Huang and G. W. J. Fleet, J. Org. Chem., 2004, 69, 997 CrossRef CAS PubMed; (d) O. David, J. Blot, C. Bellec, M.-C. Fargeau-Bellassoued, G. Haviari, J.-P. Célérier, G. Lhommet, J.-C. Gramain and D. Gardette, J. Org. Chem., 1999, 64, 3122 CrossRef CAS PubMed.
- C. Cimarelli and G. Palmieri, J. Org. Chem., 1996, 61, 5557 CrossRef CAS.
- Y. Wang, S.-P. Luo, H. Geng, Y.-P. Ruan and A.-E. Wang, Tetrahedron Lett., 2015, 56, 1255 CrossRef PubMed.
Footnotes |
| † In memory of Professor Dr S. Kan. |
| ‡ Electronic supplementary information (ESI) available: 1H NMR and 13C NMR spectra of the products 1a–1z, 1aa, 1ab. Crystallographic data in CIF of the products 1v. CCDC 1406598. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5qo00191a |
| This journal is © the Partner Organisations 2015 |