
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Intramolecular C–H insertion vs. Friedel–Crafts coupling induced by silyl cation-promoted C–F activation†
Oliver
Allemann
ab,
Kim K.
Baldridge
*ab and
Jay S.
Siegel
*ab
aDepartment of Chemistry, University of Zürich Winterthurerstrasse 190, 8057 Zürich, Switzerland
bSchool of Pharmaceutical Science and Technology, Tianjin University, 92 Weijin Road, Nankai District, Tianjin, 300072, PR China. E-mail: dean_spst@tju.edu.cn
First published on 3rd July 2015
Abstract
Silyl cation-promoted aryl C–F activation can lead to formal C–H activation and the formation of new C(ar)–C(alkyl) bonds. Deuterium-labeling experiments suggest an insertion of a phenyl cation into the C–H bond. From competition experiments, a relation between reaction rate and C–H bond strength could be established. Mechanistic parallels are drawn to the Mascarelli reaction.
Introduction
Silyl cations activate even highly stable C–F bonds.1,2 Prominent cases include hydrodefluorinations of fluoroalkanes, by simple3–5 as well as bridging6,7 cationic silanes, and catalytic intramolecular coupling of a fluoroarene with a non-functionalized arene.8,9 Specifically, triisopropylsilylium hexachloro-carba-closo-dodecaborate (1), induces the cleavage of an aryl carbon–fluorine bond allowing a proximal aryl moiety to attack the incipient phenyl cation. If attack by the adjacent arene generates too much strain, as in the formation of a 4-membered ring, the reaction is either very slow or not observed; however, in such cases the presence of an alternative nucleophile, such as the C–H sigma bond of a proximal methyl group, can create a competitive reaction path to capture the phenyl cation (Scheme 1). Exactly this C–F/C–H activation couple forms the basis of this report.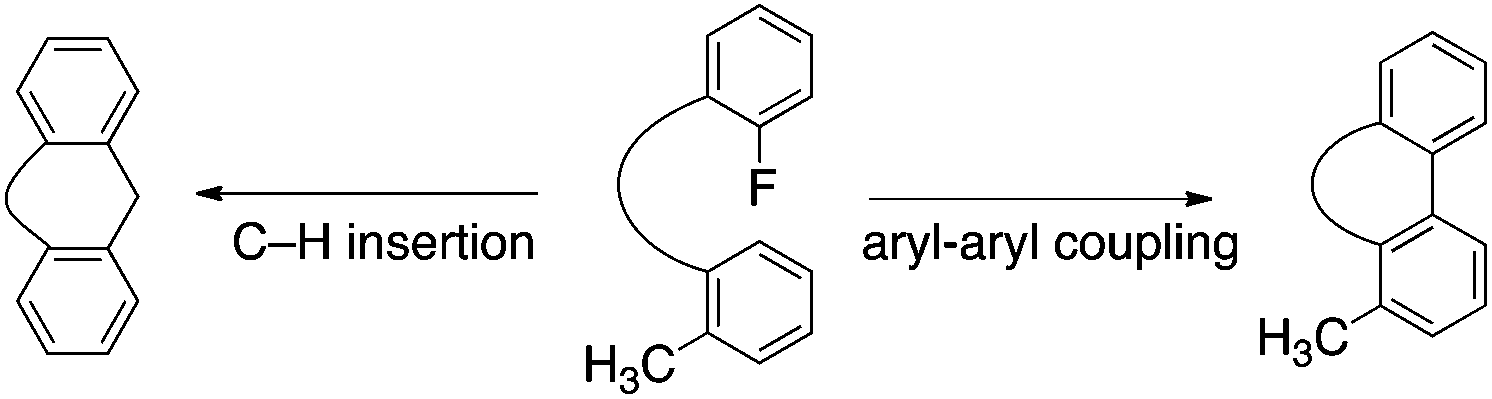 | ||
| Scheme 1 Friedel–Crafts type arylation (right) and C–H insertion, involving an alkyl group (left). | ||
Reaction of 2-fluoro-2′-methylbiphenyl (2) with 5 mol% of [iPr3Si][CHB11H5Cl6] (1) and stoichiometric dimethyldimesitylsilane (DMDMS) illustrates the fundamental C–F/C–H activation couple (Scheme 2); wherein 1 serves as catalytic initiator and DMDMS serves as Brønsted base and pro-Lewis acid catalyst. Notably, C–F activation of 2 does not lead to the highly strained biphenylene, but rather to fluorene (3).
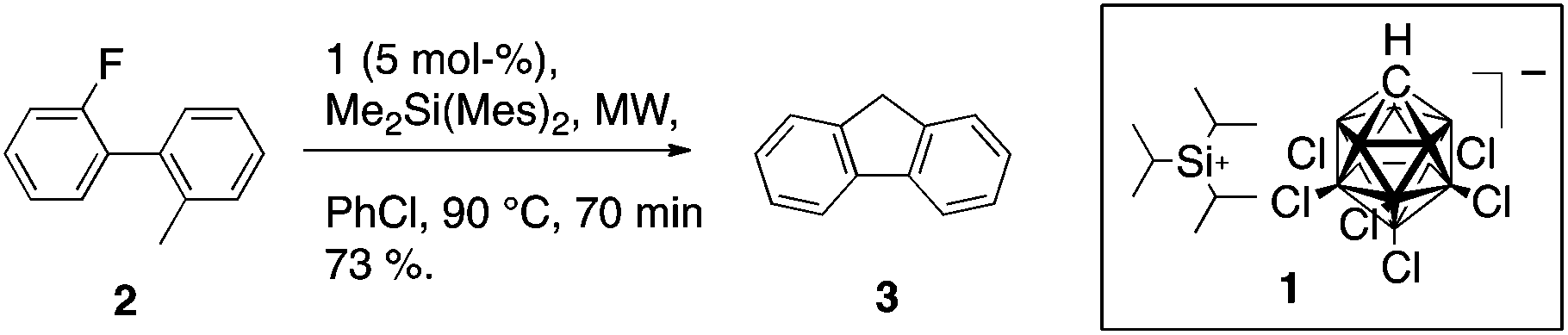 | ||
| Scheme 2 Transformation of 2-fluoro-2′-methylbiphenyl to give fluorene using silyl cation 1 as initiator. | ||
Analogous diazoniumbiphenyls exhibit similar reactivity under either thermal or acidic conditions, as observed by Mascarelli in 1932.10,11 Recently, studies on Brønsted acid-mediated intramolecular cyclization of biaryl triazenes for the synthesis of fluorenes showed similar results.12 Decomposition of these diazonium salts to form arene carbonium ions has precedence.13
Speculations on the mechanism of the Mascarelli and analogous reactions are plentiful with one of the most persistent theories being the shift of a hydride from the methyl group to the phenyl cation, resulting in a benzyl cation, followed by intramolecular electrophilic aromatic substitution (Scheme 3). Alternative mechanisms for the Mascarelli reaction include intermediates such as a pentacoordinated carbocation14 or a Zwitterion,15 postulated to be formed by abstraction of a benzylic proton; however no credible experimental evidence has been provided.
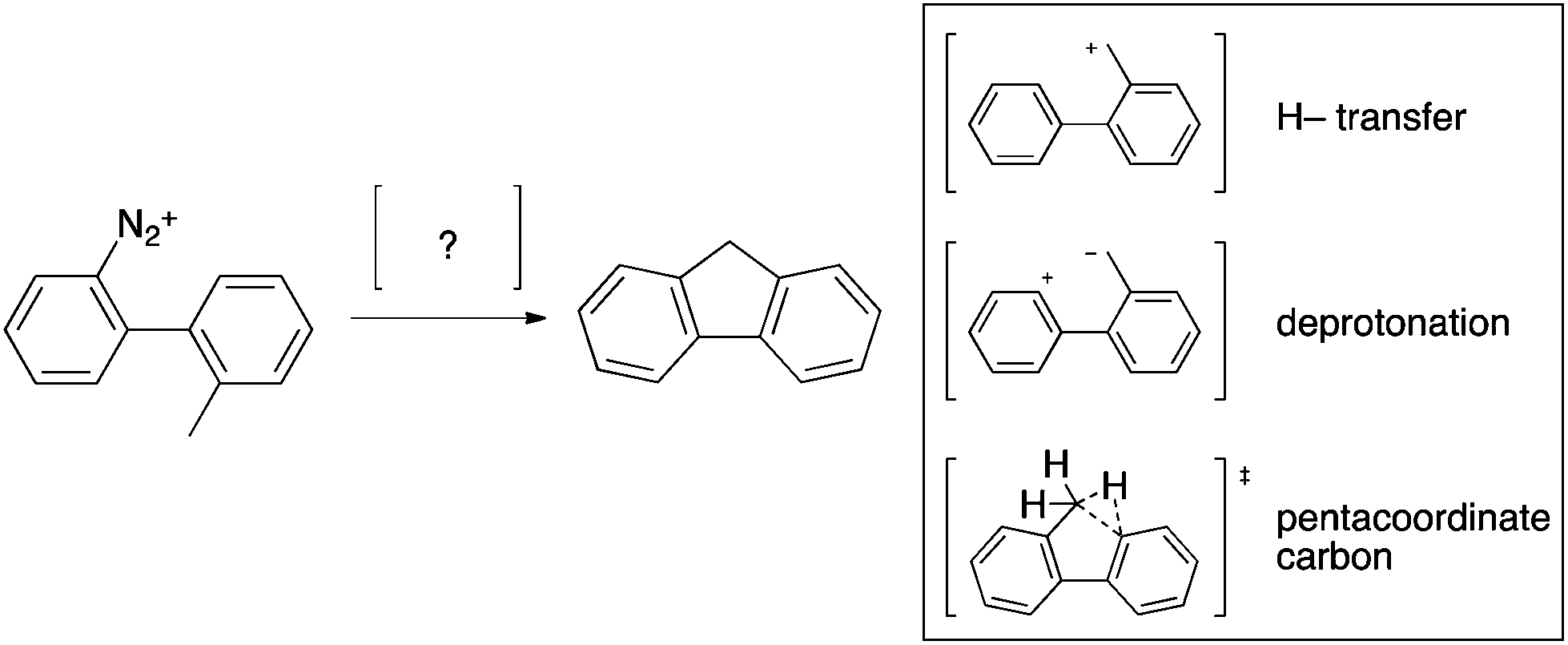 | ||
| Scheme 3 Postulated mechanisms for the Mascarelli reaction.10,11,14,15 | ||
Results and discussion
Evidence for a hydride shift was observed in the denitrogenation of 2-diazonium-N,N-dibenzylbenzamides;16 the resulting iminium ion does not undergo ring closure, but leads to products of hydrolysis. The phthalimidine originating from intramolecular cyclization, was proposed to be generated from the initial phenyl cation by an insertion mechanism.17 A hydride shift-derived mechanism was further inferred from the observation of a 2-phenylbenzyl alcohol, generated likely from the reaction of the 2-phenylbenzyl cation with water;18 but, only 8% of alcohol was isolated. Notably, the benzyl cation generated by protonation of 2-biphenylmethanol with Amberlyst-15 in benzene showed only coupling with solvent;14 and, heating in cyclohexane produced fluorene, but very slowly compared to the known rate of the Mascarelli and related reactions.Thermal decomposition of the 2′-trifluoromethylbiphenyl-2-yldiazonium salt generates a difluorobenzylcation, which cyclizes in hexafluorobenzene under oxygen atmosphere.19 An example relying on niobium-mediated C–F activation was also reported, where 2-trifluoromethylbiphenyl was transformed to 9,9-difluorofluorene.20,21
On the basis of the assumption that silylium-activated fluoroarenes react in a similar fashion as diazonium compounds, the deuterated analog 2-d3 was synthesized to observe the fate of the methyl hydrogens. MS and NMR analysis showed that most of the deuterium atoms were still present in the product (ca. 85%). The deuterium distribution within the product was determined by 2H and 1H NMR spectroscopy to be 60% @ C1, 18% @ C2, 2% @ C3, 5% @ C4, and ca. 15% deuterium loss.22 Thus, products from deuteration at C1, or C2, plus deuterium loss account for 90+ % of the product formation. Very little deuterium ends up at C4, inconsistent with the expectation of a hydride abstraction mechanism. In contrast, an insertion of the phenyl cation into the carbon deuterium bond followed by elimination or migration could account for this product distribution by the following rationale (Scheme 4).
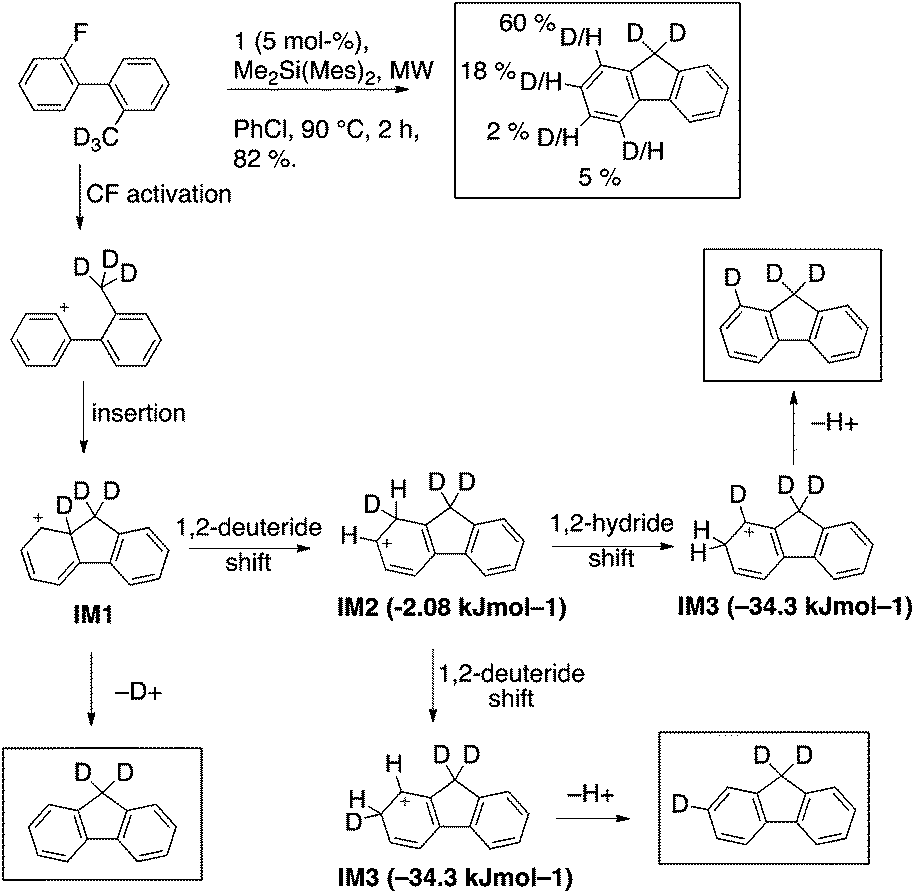 | ||
| Scheme 4 Deuterium distribution in 3 produced from 2-d3; postulated intermediates and their M06-2X/Def2-TZVPP(chlorobenzene) computed energetics. | ||
Intermediate 1, IM1, (a Wheland cation) can either eliminate to yield fluorene possessing no deuterium atom at the arene, or undergo a deuteride shift to form intermediate 2, IM2. IM2 could rapidly transform by another migration into the much more stable intermediate 3, IM3, from which the majority of elimination occurs. Taking into account that eliminations and migrations occur more readily for hydrogen than deuterium, one would predict a pronounced deuteration at position 1, but also substantial deuteration at position 2.
The stabilities of IM1 and IM2 were calculated to be very similar (ΔE(IM2–IM1) = −2.1 kJ mol−1), in contrast to IM3, which is much lower in energy (ΔE(IM3–IM1) = −34.3 kJ mol−1).23 Transition state TS(1,2), is calculated to be rate determining over TS(2,3) [ETS(1,2) = 25.7 kJ mol−1vs. ETS(2,3) = 21.7 kJ mol−1] for the hydride migration IM1–IM2–IM3. This computational data leads to a model in which the initially formed IM1 partitions with preference for intramolecular migration over abstraction of D+. IM2 is fleeting and further hydride migration occurs to IM3 without appreciable elimination. A Hammond Postulate analysis allows this to be modeled by basic principles of physical organic chemistry (Scheme 5).
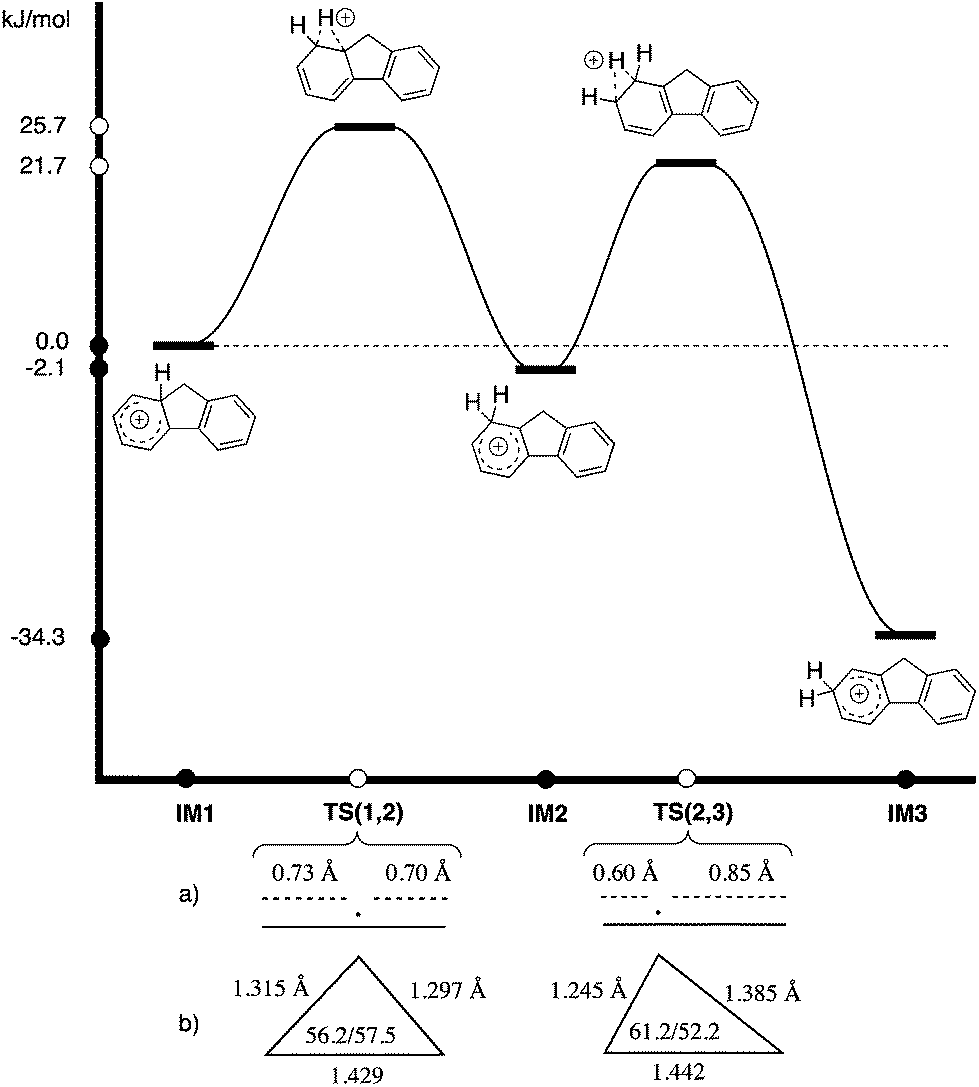 | ||
| Scheme 5 Calculated reaction energy profile (M06-2X/Def2-TZVPP in chlorobenzene)23 for hydride migration from IM1–IM2–IM3 [TS(1,2) rate determining]. (a) Projection of hydride position in TS(1,2) and TS(2,3) onto the carbon–carbon bond length as an estimate of late/early character; (b) triangle defined by carbon and hydrogen positions in TS(1,2) and TS(2,3) to show late/early TS assignment. | ||
Intramolecular hydride transfer to form a benzyl cation followed by ring closure would place substantial deuteration at C4, which is not observed. Additionally, abstraction of fluoride from 2-fluoromethylbiphenyl (4) yielded primarily 5 and 6 (coupling with mesitylene from DMDMS24) and traces of 7 but no fluorene, further demonstrating the preference of the benzyl cation for intermolecular coupling (Scheme 6). Only trace amounts of two intermolecular coupling isomers with DMDMS could be observed for the reaction of 2. Thus, intramolecular hydride transfer seems mechanistically insignificant in the formation of fluorene from 2.
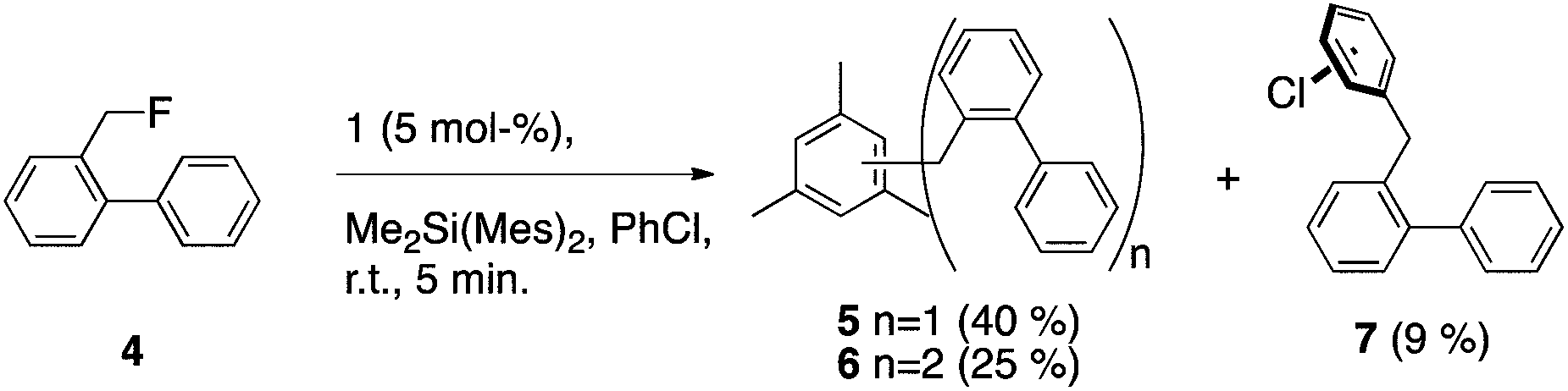 | ||
| Scheme 6 Product distribution from abstraction of fluoride from 4. | ||
C–F activation of 2 led exclusively to 3 by benzyl C–H insertion. In contrast, for substrates 8a–8c where benzyl C–H insertion and Friedel–Crafts reaction pathways are probable, a competitive distribution of products can be observed (Scheme 7). Whereas the substrate possessing the stronger benzylic C–H bond (8a, toluene-like: 375 kJ mol−1) prefers formation of the fluoranthene derivative 9a over 10a, with a ratio of 1.6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.0, the substrates featuring the weaker benzylic C–H bond (8b & 8c, cumene-like: 348 kJ mol−1) rather form products of the insertion 10b and 10c (1.0:1.9 and 1.0:2.1, respectively).
:1.0, the substrates featuring the weaker benzylic C–H bond (8b & 8c, cumene-like: 348 kJ mol−1) rather form products of the insertion 10b and 10c (1.0:1.9 and 1.0:2.1, respectively).
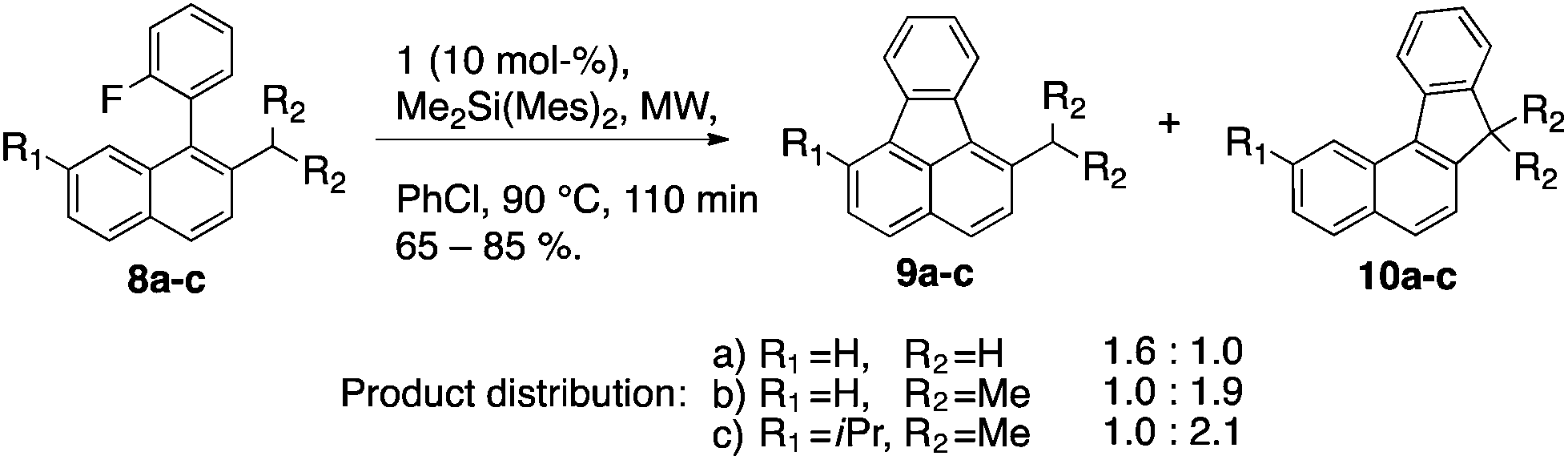 | ||
| Scheme 7 Product distributions from C–F activation 8a–c. | ||
Computational studies predict an activation energy for C–F activation of ∼100 kJ mol−1, which is consistent with the reaction proceeding at 90 °C over 2 h. Calculated transition states leading to products 9a and 10a are very close in energy (ΔETS(9a–10a) = −1.2 kJ mol−1 [ε = 5.7 chlorobenzene]).15 Experimental product ratios also suggest that the transition states are very close in energy (ΔETS((9a–10a) = +1.5 kJ mol−1). The difference between theory and experiment (2.7 kJ mol−1 in chlorobenzene) is well within the limits of reliability – emphasizing the importance of small energy differences for product ratios in the range of 1:2 to 2:1 (ca. 4 kJ mol−1). Computations suggest that changes in dielectric constant can influence the product selectivity (ΔETS(9a–10a) = +1.5 kJ mol−1 [ε = 1 vacuum]) but, they do not change the general course and nature of the reaction.
Noteworthy is that only a small fraction of the difference in C–H bond strength shows up in the difference in transition state energy (ca. 5–10%). Invoking the Hammond postulate again here, one would conclude that the reaction product selectivity step has an early transition state, further supporting a direct C–H insertion mechanism. An early C–H insertion mechanism would predict the reaction to be relative promiscuous with regard to C–H insertion chemistry. Indeed, the reactivity is not restricted to weak benzylic CH groups. Applying the reaction conditions on 2-fluoro-2′-tert-butylbiphenyl (11) also yields ring-closed product (12) (Scheme 8). Despite the large difference in C–H bond strength for a primary aliphatic CH3 group compared to a benzylic CH3 group (ΔE ∼40–50 kJ mol−1), the formation of the six-membered ring, aliphatic insertion product, is only slightly slower than that of the model compound.25 This result further supports the early transition state, direct insertion model for this reaction.
 | ||
| Scheme 8 Non-benzylic C–H bond insertion from C–F activation of 11. | ||
Conclusions and outlook
Parallel to the Mascarelli reaction for diazonium salts, phenyl cations generated from arylfluorides by C–F bond activation with cationic silyl Lewis acids can undergo intramolecular arene-coupling and C–H insertion. Deuterium labeling studies suggest a direct C–H insertion mechanism. Competition experiments showed only a slight dependence of aryl–aryl coupling vs. C–H insertion based on the C–H bond strength, consistent with an early-transition-state to a direct-insertion mechanism. The tandem activation of two generally stable functional groups (Ar–F, alkyl–H) provides a new reactivity pattern in cationic silyl Lewis acids.Experimental and computational details
Synthesis and characterization data, together with computational details, are available in the ESI.†Acknowledgements
We thank the Swiss National Science Foundation, the National Basic Research Program of China (2015CB856500), the Qian Ren Scholar Program of China, and the Synergetic Innovation Center of Chemical Science and Engineering (Tianjin), for support of this work.Notes and references
- T. Stahl, F. T. Klare and M. Oestreich, ACS Catal., 2013, 3, 1578–1587 CrossRef CAS.
- H. Amii and K. Uneyama, Chem. Rev., 2009, 109, 2119 CrossRef CAS PubMed.
- V. J. Scott, R. Çelenligil-Çetin and O. V. Ozerov, J. Am. Chem. Soc., 2005, 127, 2852–2853 CrossRef CAS PubMed.
- C. Douvris and O. V. Ozerov, Science, 2008, 321, 5893 CrossRef PubMed.
- C. Douvris, C. M. Nagaraja, C. H. Chen, B. M. Foxman and O. V. Ozerov, J. Am. Chem. Soc., 2010, 24, 533 Search PubMed.
- R. Panisch, M. Bolte and T. Müller, J. Am. Chem. Soc., 2006, 128, 9676–9682 CrossRef CAS PubMed.
- N. Luhmann, R. Panisch and T. Müller, Appl. Organomet. Chem., 2010, 24, 533 CrossRef PubMed.
- S. Duttwyler, C. Douvris, N. L. P. Fackler, F. S. Tham, C. A. Reed, K. K. Baldridge and J. S. Siegel, Angew. Chem., Int. Ed., 2010, 49, 7519 CrossRef CAS PubMed.
- O. Alleman, S. Duttwyler, P. Romanato, K. K. Baldridge and J. S. Siegel, Science, 2011, 332, 574–577 CrossRef PubMed.
- L. Mascarelli, Gazz. Chim. Ital., 1936, 66, 843 CAS.
- L. Mascarelli, in L'industria Chim., Torino, 1932, p. 910 Search PubMed.
- L. Xu, W. Yang, L. Zhang, M. Miao, Z. Yang, X. Xu and H. Ren, J. Org. Chem., 2014, 79, 9206 CrossRef CAS PubMed.
- J. F. Chlobowski, Carbonium Ions, Wiley-Interscience, New York, 1970 Search PubMed.
- A. S. Daudpota and H. Heaney, Tetrahedron Lett., 1978, 19, 3471 CrossRef.
- I. Puskas and E. K. Fields, J. Org. Chem., 1968, 33, 4237 CrossRef CAS.
- T. Cohen and J. Lipowitz, J. Am. Chem. Soc., 1964, 86, 2514 CrossRef CAS.
- T. Cohen and J. Lipowitz, J. Am. Chem. Soc., 1964, 86, 2515 CrossRef CAS.
- R. W. Stumpe, Tetrahedron Lett., 1980, 21, 4891 CrossRef CAS.
- D. Ferraris, C. Cox, R. Anand and T. Lectka, J. Am. Chem. Soc., 1997, 119, 4319 CrossRef CAS.
- K. Fuchibe, K. Mitomi, R. Suzuki and T. Akiyama, Chem. – Asian J., 2008, 3, 261 CrossRef CAS PubMed.
- K. Fuchibe and T. Akiyama, J. Am. Chem. Soc., 2006, 128 Search PubMed.
- The deuterium distribution was determined by using both 1H and 2H NMR spectroscopic data.
- Additional details concerning the computations can be found in the ESI.† As noted by one referee, solvent caged species may be present in these reactions, but they do not seem to steer the course of reactivity.
- Also double addition to mesitylene could be observed. A similar reactivity was reported by Müller et al.7.
- W. M. Haynes, CRC Handbook of Chemistry and Physics, CRC Press, Boca Raton, FL, 2012 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5qo00170f |
| This journal is © the Partner Organisations 2015 |