
Engineering ligands on the Au center: discovering broadly applicable gold catalysis with high turnover numbers
Suleman M.
Inamdar
and
Nitin T.
Patil
*
Organic Chemistry Division, CSIR-National Chemical Laboratory, Dr Homi Bhabha Road, Pune - 411 008, India. E-mail: n.patil@ncl.res.in; Web: http://academic.ncl.res.in/n.patil/home Fax: (+) 91-20-25902629
First published on 12th June 2015
Abstract
The research groups of Hammond/Xu and Zhang report the discovery of new ligands for gold catalysis. With these ligands, gold catalyzed reactions have been reported for a broader range of substrates with very high TONs at a level that can match the needs of large-scale applications.
 Suleman M. Inamdar | Suleman M. Inamdar received his MSc degree in organic chemistry from the University of Pune in 2001. He joined CSIR-NCL Pune, as a Project assistant-II and worked for about two years in OCT division. Later, in May 2004 he moved to Aditya Birla Science & Technology Centre, Mumbai and worked for about five years in R & D division as a Sr. Research Associate. He has been awarded as “Young Professional-2007” a prestigious award from the Aditya Birla group at Bangkok. He completed his doctoral study at CSIR-NCL affiliated to the University of Pune in 2013. Presently he is working as a Postdoctoral fellow in Dr Nitin T. Patil's research group at CSIR-NCL Pune. His major research interest includes the development of metal–organo catalyzed enantioselective reactions and their application for the total synthesis of bioactive molecules. |
 Nitin T. Patil | Nitin T. Patil was born in Jalgaon (Maharashtra), India, in 1975. He completed his doctoral study at the University of Pune in 2002 under the supervision of Prof. D. D. Dhavale. After working as a postdoc at Goettingen with Professor Christoph Schneider, he moved to Tohoku University, Japan, as a JSPS fellow. Later, in April 2005, he was appointed as Assistant Professor in Prof. Yoshinori Yamamoto's laboratory. In June 2006, he joined Prof. K. C. Nicolaou's laboratory (PI: Prof. David Chen) at Singapore, and later at The Scripps Research Institute, USA. He began his independent career in September 2008 at IICT, Hyderabad, India. Recently, he has moved to CSIR-NCL, Pune in August 2013. He has been the recipient of INSA Young Scientist Medal-2010 and Alkyl Amines-ICT Foundation Day Young Scientist Award-2010. He has also been elected as “Young Associate” of the Indian Academy of Sciences, Bangalore in 2010. His broad research interests include the development of metal–, organo– and metaloorgano catalyzed enantioselective methods and total synthesis of natural products. |
The development of homogeneous gold catalysis in the past decade has been remarkably rapid and the field now is considered as a landmark addition to the field of organic synthesis.1 In most cases, gold catalysis involves the coordination of a C–C multiple bond to the metal catalyst thereby enhancing its electrophilicity for the subsequent attack of nucleophiles. The fact is that gold catalysts are expensive and the common drawback associated with homogeneous gold catalysis is the difficulty in catalyst recovery. A literature survey revealed that most of the reactions based on homogeneous gold catalysis require >0.5 mol% of catalyst loading. These facts continuously reinforced the existing perception that gold catalysis would be too expensive to be useful for industrial processes.
One of the greatest challenges that an organic chemist faces today is to develop economically viable gold chemistry. While there is a little success on the use of heterogeneous gold catalysis,2 the reactions have been reported for a narrow set of substrates and catalyst leaching is one of the major obstacles. Because of the obvious advantages of homogeneous catalysis, the “ligand design” strategy which is fairly common in homogeneous transition metal catalysis has to be applied in gold catalysis. This would enable one to achieve high catalyst turnover numbers (TONs) enabling the gold chemistry to find a place in large-scale synthesis and expanding the horizon of gold catalysis from academia to industry.
Indeed, various researchers across the globe realized the importance of this field and reported the catalyst loadings in the ppm range.3 However, successful examples are limited and reactions are specific. Until recently, there has been no general catalyst available which catalyzes a large variety of reactions with an extremely low catalyst loading. Very recently, the research group of Hammond/Xu4 and Zhang5 has independently reported the rational design and synthesis of novel ligands for gold catalysis – the catalytic system is highly general and works with an extremely low catalyst loading.
The ligand design1ak by Hammond and Xu's research group focused on three major factors that account for the high loading of gold catalysts (Scheme 1).4 These factors are: (1) the decay of the gold catalyst during the reaction; (2) the mismatch of electronic effects within the gold ligand; and (3) the formation of off-cycle gold species. Keeping in mind the above factors, the authors systematically investigated the effect of various phosphine ligands on the Au center for intermolecular hydroamination of phenyl acetylene with aniline. They found that the reaction with Ph3P was very slow; while the reaction with JohnPhos, generated a product with a much higher rate with full conversion. The low reaction rate, in the case of PPh3, could have been caused by the electronic effect mismatch in the ligand, as this reaction needs an electron-rich ligand. Furthermore, the gradual deactivation of the catalyst contributed to the low rate of the reaction. These experiments indicated the importance of ortho substitution and electronic density matching in the ligand.6 The authors realized that: (1) the proximity of the o-phenyl to the gold center prevents the deactivation of gold(I) (refer I), (2) the turnover limiting stage in the majority of gold-catalyzed reactions is the regeneration of the cationic gold catalyst from the gold σ-complex intermediate (refer I) via protodeauration.6 With this logic, it is expected that an electron-rich ligand capable of supplying the electronic density to the gold metal center would facilitate the regeneration of the cationic gold catalyst. The ligand design by Hammond and Xu also considers the observation reported by Widenhoefer that the off-cycle gold species bis-Au-vinyl species II is responsible for reducing the turnover.7 It was envisaged that the two sterically demanding biaryls on the phosphine ligand could surround the gold center and discourage the formation of II. Thus, the research group of Hammond and Xu designed a phosphine ligand L-1 that contains two electron-rich o-biphenyl groups and a cyclohexyl substituent. As can be judged from Scheme 1, the ligand L-1 formed a gold complex that catalyzed some common types of gold-catalyzed reactions including intra- and intermolecular X–H additions to alkynes (A–F and H) and cycloisomerizations (G, I and J), with high turnover numbers at room temperature or slightly elevated temperatures. We noted that the catalyst (L-1)·AuCl is made available by Aldrich under the name BisPhePhos XD gold(I) chloride.
 | ||
| Scheme 1 Gold catalysis with (L-1)·AuX. | ||
Soon after, Zhang et al. demonstrated a ligand design that offers a conceptually new and potentially general approach to achieve highly efficient gold catalysis with ultra-low catalyst loadings (Scheme 2).5 The authors envisioned that a ligand with a rigid and extending framework could project a functional group (FG) farther enough to reach an approaching nucleophile (L-2). The ligand was designed in such a way that it could have H-bonding interaction with the nucleophile and hence render the attack of an incoming nucleophile at the gold-activated triple bond in an intramolecular event (refer III). The rigidity of the backbone would minimize the entropy cost for organizing transition states, and the directing group should be flexible enough to accommodate a broad substrate scope. This strategy is expected to facilitate the initial nucleophilic attack drastically by converting it from an intermolecular process to a quasi-intramolecular event and may also accelerate subsequent transformations. As a result, higher catalyst TONs and hence more efficient catalysis could be expected.
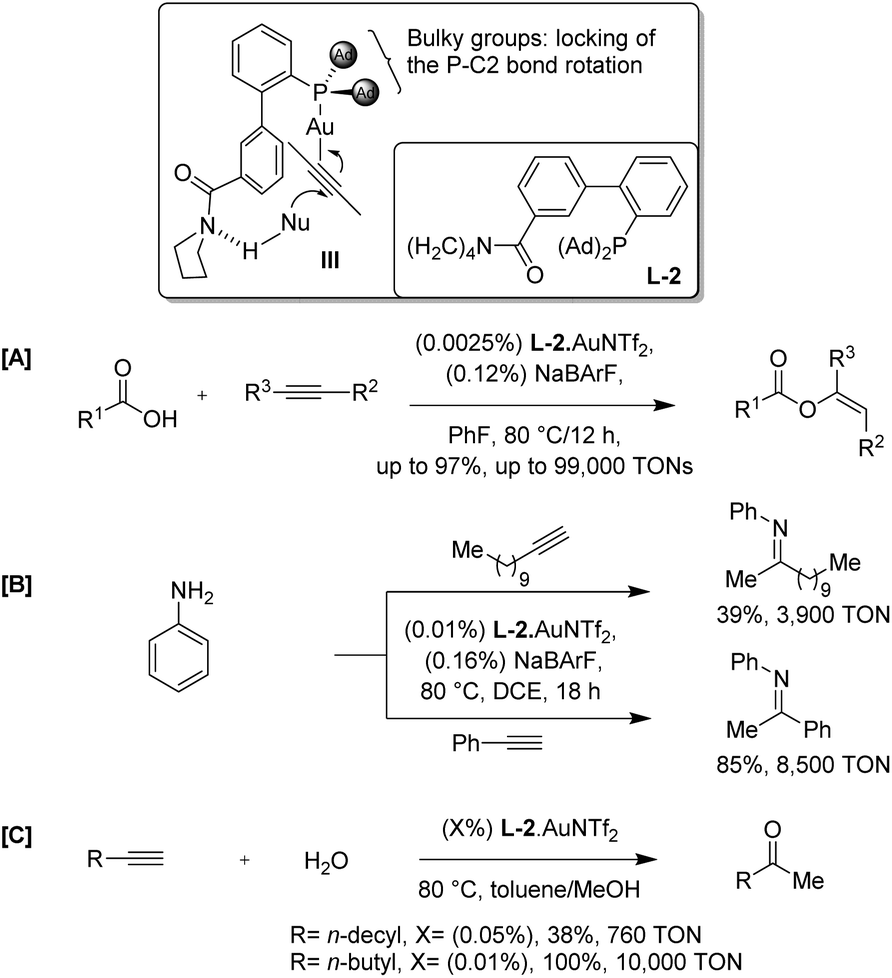 | ||
| Scheme 2 Gold catalysis with (L-2)·AuNTf2. | ||
They discovered that the gold complex, (L-2)·AuNTf2, catalyzes the addition of carboxylic acids to alkynes with catalyst TONs up to 99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 (Scheme 2A). Density functional theory calculations support the role of the amide moiety in directing the attack of carboxylic acid via hydrogen bonding. The general applicability of the design in other gold catalyzed reactions is demonstrated by highly efficient hydroamination and hydration of alkynes. In the case of hydroamination of 1-dodecyne with aniline using (L-2)·AuNTf2, catalyst TONs reached 3900; while, for phenyl acetylene they reached 8500 (Scheme 2B). The complex, (L-2)·AuNTf2, also facilitate the nucleophilic attack by H2O, resulting in substantially accelerated hydration of alkynes in methanol with 100 ppm of the catalyst and TONs of at least 10000 with a quantitative yield (Scheme 2C).
000 (Scheme 2A). Density functional theory calculations support the role of the amide moiety in directing the attack of carboxylic acid via hydrogen bonding. The general applicability of the design in other gold catalyzed reactions is demonstrated by highly efficient hydroamination and hydration of alkynes. In the case of hydroamination of 1-dodecyne with aniline using (L-2)·AuNTf2, catalyst TONs reached 3900; while, for phenyl acetylene they reached 8500 (Scheme 2B). The complex, (L-2)·AuNTf2, also facilitate the nucleophilic attack by H2O, resulting in substantially accelerated hydration of alkynes in methanol with 100 ppm of the catalyst and TONs of at least 10000 with a quantitative yield (Scheme 2C).
Though the field of homogeneous catalysis has flourished tremendously, the reports dealing with the discovery of efficient catalytic systems are relatively few. The lack of a general catalyst system which works over a broad range of substrates with high TONs, restricts the use of this chemistry for industrial applications in the synthesis of bulk chemicals. The reports by Hammond/Xu and Zhang described in this highlight, report the discovery of new ligands for gold catalysis. With these ligands, gold catalyzed reactions have been reported for a broader range of substrates with very high TONs at a level that can match the needs of medium- or large-scale applications.
A research by Hammond/Xu and Zhang would likely inspire others to search for new ligands which are highly efficient for gold-catalyzed transformations. The results will open the door for future industrial applications in the synthesis of fine chemicals. Though, this highlight has shown that a low catalyst loading is possible to be achieved, all the reactions produce racemic compounds. Nothing is known about chiral gold catalysis with high TONs to generate enantio-pure compounds for real practical applications. Next challenges for this chemistry could include the development of bifunctional chiral catalysts, bearing phosphine and directing groups, for an enantioselective gold catalyzed reaction with a very low catalyst loading.
Acknowledgements
We gratefully acknowledge financial support by the Department of Science and Technology (No. SB/S1/OC-17/2013), India for our research in the related area.Notes and references
- Selected reviews: (a) B. Ranieri, I. Escofet and A. M. Echavarren, Org. Biomol. Chem., 2015 10.1039/c5ob00736d; (b) R. Dorel and A. M. Echavarren, Chem. Rev., 2015 DOI:10.1021/cr500691k; (c) J. A. Goodwin and A. Aponick, Chem. Commun., 2015, 51, 8730 RSC; (d) D. Qiana and J. Zhang, Chem. Soc. Rev., 2015, 44, 677 RSC; (e) V. K.-Y. Lo, A. O.-Y. Chan and C.-M. Che, Org. Biomol. Chem., 2015, 13, 6667 RSC; (f) Y. Wang, M. E. Muratore and A. M. Echavarren, Chem. – Eur. J., 2015, 21, 7332 CrossRef CAS PubMed; (g) M. Jia and M. Bandin, ACS Catal., 2015, 5, 1638 CrossRef CAS; (h) A. S. K. Hashmi, Acc. Chem. Res., 2014, 47, 864 CrossRef CAS PubMed; (i) Y.-M. Wang, A. D. Lackner and F. D. Toste, Acc. Chem. Res., 2014, 47, 889 CrossRef CAS PubMed; (j) A. Fürstner, Acc. Chem. Res., 2014, 47, 925 CrossRef PubMed; (k) L. Fensterbank and M. Malacria, Acc. Chem. Res., 2014, 47, 953 CrossRef CAS PubMed; (l) S. M. Inamdar, A. Konala and N. T. Patil, Chem. Commun., 2014, 50, 15124 RSC; (m) J. Xie, C. Pan, A. Abdukader and C. Zhu, Chem. Soc. Rev., 2014, 43, 5245 RSC; (n) W. Yang and A. S. K. Hashmi, Chem. Soc. Rev., 2014, 43, 2941 RSC; (o) M. E. Muratore, A. Homs, C. Obradors and A. M. Echavarren, Chem. – Asian J., 2014, 9, 3066 CrossRef CAS PubMed; (p) L. Zhang, Acc. Chem. Res., 2014, 47, 877 CrossRef CAS PubMed; (q) C. Obradors and A. M. Echavarren, Acc. Chem. Res., 2014, 47, 902 CrossRef CAS PubMed; (r) T. Cañeque, F. M. Truscott, R. Rodriguez, G. Maestri and M. Malacria, Chem. Soc. Rev., 2014, 43, 2916 RSC; (s) I. Braun, A. M. Asiri and A. S. K. Hashmi, ACS Catal., 2013, 3, 1902 CrossRef CAS; (t) W. E. Brenzovich Jr., Angew. Chem., Int. Ed., 2012, 51, 8933 CrossRef PubMed; (u) D. Garayalde and C. Nevado, ACS Catal., 2012, 2, 1462 CrossRef CAS; (v) B.-L. Lu, L. Dai and M. Shi, Chem. Soc. Rev., 2012, 41, 3318 RSC; (w) N. T. Patil, Chem. – Asian J., 2012, 7, 2186 CrossRef CAS PubMed; (x) M. Bandini, Chem. Soc. Rev., 2011, 40, 1358 RSC; (y) B. Biannic and A. Aponick, Eur. J. Org. Chem., 2011, 6605 CrossRef CAS PubMed; (z) A. Corma, A. Leyva-Pérez and M. J. Sabater, Chem. Rev., 2011, 111, 1657 CrossRef CAS PubMed; (a a) H. A. Wegner and M. Auzias, Angew. Chem., Int. Ed., 2011, 50, 8236 CrossRef CAS PubMed; (a b) A. S. Dudnik, N. Chernyak and V. Gevorgyan, Aldrichimica Acta, 2010, 43, 37 CAS; (a c) S. Sengupta and X. Shi, ChemCatChem, 2010, 2, 609 CrossRef CAS PubMed; (a d) N. T. Patil, R. D. Kavthe and Y. Yamamoto, Adv. Heterocycl. Chem., 2010, 101, 75 CAS; (a e) A. S. K. Hashmi, Angew. Chem., Int. Ed., 2010, 49, 5232 CrossRef CAS PubMed; (a f) S. Ma, Handbook of Cyclization Reactions, Wiley-VCH, 2010, ch. 10 Search PubMed; (a g) A. S. K. Hashmi and M. Bührle, Aldrichimica Acta, 2010, 43, 27 CAS; (a h) P. Garcia, M. Malacria, C. Aubert, V. Gandon and L. Fensterbank, ChemCatChem, 2010, 2, 493 CrossRef CAS PubMed; (a i) S. M. Abu Sohel and R.-S. Liu, Chem. Soc. Rev., 2009, 38, 2269 RSC; (a j) A. Fürstner, Chem. Soc. Rev., 2009, 38, 3208 RSC; (a k) D. J. Gorin, B. D. Sherry and F. D. Toste, Chem. Rev., 2008, 108, 3351 CrossRef CAS PubMed; (a l) Z. Li, C. Brouwer and C. He, Chem. Rev., 2008, 108, 3239 CrossRef CAS PubMed; (a m) A. Arcadi, Chem. Rev., 2008, 108, 3266 CrossRef CAS PubMed; (a n) E. Jiménez-Núñez and A. M. Echavarren, Chem. Rev., 2008, 108, 3326 CrossRef PubMed; (a o) A. S. K. Hashmi and M. Rudolph, Chem. Soc. Rev., 2008, 37, 1766 RSC; (a p) A. Fürstner and P. W. Davies, Angew. Chem., Int. Ed., 2007, 46, 3410 CrossRef PubMed; (a q) A. S. K. Hashmi, Chem. Rev., 2007, 107, 3180 CrossRef CAS PubMed; (a r) E. Jiménez-Núñez and A. M. Echavarren, Chem. Commun., 2007, 333 RSC; (a s) L. Zhang, J. Sun and S. A. Kozmin, Adv. Synth. Catal., 2006, 348, 2271 CrossRef CAS PubMed; (a t) A. S. K. Hashmi and G. J. Hutchings, Angew. Chem., Int. Ed., 2006, 45, 7896 CrossRef PubMed; (a u) R. A. Widenhoefer and X. Han, Eur. J. Org. Chem., 2006, 4555 CrossRef CAS PubMed; (a v) S. Ma, S. Yu and Z. Gu, Angew. Chem., Int. Ed., 2006, 45, 200 CrossRef CAS PubMed.
- Reviews: (a) N. T. Patil, ChemCatChem, 2011, 3, 1121 CrossRef CAS PubMed; (b) A. Corma and H. Garcia, Chem. Soc. Rev., 2008, 37, 2096 RSC. Selected references: (c) F. Schröder, M. Ojeda, N. Erdmann, J. Jacobs, R. Luque, T. Noël, L. Van Meervelt, J. Van der Eycken and E. V. Van der Eycken, Green Chem., 2015, 17, 3314 RSC; (d) I. Cano, A. M. Chapman, A. Urakawa and P. W. N. M. van Leeuwen, J. Am. Chem. Soc., 2014, 136, 2520 CrossRef CAS PubMed; (e) N. Dupré, C. Brazel, L. Fensterbank, M. Malacria, S. Thorimbert, B. Hasenknopf and E. Lacôte, Chem. – Eur. J., 2012, 18, 12962 CrossRef PubMed; (f) C. Efe, I. N. Lykakis and M. Stratakis, Chem. Commun., 2011, 47, 803 RSC; (g) M. García-Mota, N. Cabello, F. Maseras, A. M. Echavarren, J. Pérez-Ramírez and N. Lopez, ChemPhysChem, 2008, 9, 1624 CrossRef PubMed; (h) F. Neatu, Z. Li, R. Richards, P. Y. Toullec, J.-P. Genêt, K. Dumbuya, J. M. Gottfriend, H.-P. Steinrück, V. I. Pârvulescu and V. Michelet, Chem. – Eur. J., 2008, 14, 9412 CrossRef CAS PubMed; (i) A. Corma, P. Concepción, I. Domnguez, V. Fornés and M. J. Sabater, J. Catal., 2007, 251, 39 CrossRef CAS PubMed; (j) A. Abad, A. Corma and H. García, Top. Catal., 2007, 44, 237 CrossRef CAS PubMed; (k) D. Lantosa, M. Contelb, A. Larreac, D. Szabo and I. T. Horváth, QSAR Comb. Sci., 2006, 25, 719 CrossRef PubMed; (l) S. Carrettin, M. C. Blanco, A. Corma and A. S. K. Hashmi, Adv. Synth. Catal., 2006, 348, 1283 CrossRef CAS PubMed.
- (a) M. C. Blanco Jaimes, C. R. N. Böhling, J. M. Serrano-Becerra and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2013, 52, 7963 CrossRef CAS PubMed; (b) V. Lavallo, J. H. Wright, F. S. Tham and S. Quinlivan, Angew. Chem., Int. Ed., 2013, 52, 3172 CrossRef CAS PubMed; (c) X.-F. Tu and L.-Z. Gong, Angew. Chem., Int. Ed., 2012, 51, 11346 CrossRef CAS PubMed; (d) J. Oliver-Meseguer, J. R. Cabrero-Antonino, I. Domínguez, A. Leyva-Pérez and A. Corma, Science, 2012, 338, 1452 CrossRef CAS PubMed; (e) A. S. K. Hashmi, Science, 2012, 338, 1434 CrossRef CAS PubMed; (f) M. Bouhrara, E. Jeanneau, L. Veyre, C. Copret and C. Thieuleux, Dalton Trans., 2011, 40, 2995 RSC; (g) N. Marion, R. S. Ramon and S. P. Nolan, J. Am. Chem. Soc., 2009, 131, 448 CrossRef CAS PubMed; (h) M. Comotti, C. D. Pina, R. Matarrese and M. Rossi, Angew. Chem., Int. Ed., 2004, 43, 5812 CrossRef CAS PubMed; (i) E. Mizushima, K. Sato, T. Hayashi and M. Tanaka, Angew. Chem., Int. Ed., 2002, 41, 4563 CrossRef CAS; (j) E. Mizushima, T. Hayashi and M. Tanaka, Org. Lett., 2003, 5, 3349 CrossRef CAS PubMed; (k) J. H. Teles, S. Brode and M. Chabanas, Angew. Chem., Int. Ed., 1998, 37, 1415 CrossRef CAS.
- D. Malhotra, M. S. Mashuta, G. B. Hammond and B. Xu, Angew. Chem., Int. Ed., 2014, 53, 4456 CrossRef CAS PubMed.
- Y. Wang, Z. Wang, Y. Li, G. Wu, Z. Cao and L. Zhang, Nat. Commun., 2014, 5, 3470 Search PubMed.
- W. Wang, G. B. Hammond and B. Xu, J. Am. Chem. Soc., 2012, 134, 5697 CrossRef CAS PubMed.
- T. J. Brown, D. Weber, M. R. Gagné and R. A. Widenhoefer, J. Am. Chem. Soc., 2012, 134, 9134 CrossRef CAS PubMed.
| This journal is © the Partner Organisations 2015 |