
Palladium-catalyzed intramolecular rearrangement of vinylidenecyclopropanes through C–C bond activation†
Dong
Pan
,
Gen-Qiang
Chen
,
Xiang-Ying
Tang
and
Min
Shi
*
State Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 354 Fenglin Road, Shanghai 200032, P. R. China. E-mail: mshi@mail.sioc.ac.cn
First published on 29th April 2015
Abstract
Vinylidenecyclopropanes bearing sulfonamide can undergo a novel intramolecular rearrangement to give the corresponding functionalized dimethylenecyclopropanes in moderate to good yields in the presence of Pd(OAc)2 in toluene upon heating through C–C bond activation based on weak coordination of the sulfonamide directing group. The reaction pathway can be changed for phenyl substituted vinylidenecyclopropane, giving another type of dimethylenecyclopropane in methanol in the presence of K2CO3 under reflux.
The use of coordinating moieties as directing groups for the C–H bond activation has become a powerful established method to enhance reactivity and regioselectivity.1 In this very active research arena, weak coordination as a powerful means for developing broadly useful C–H functionalization reactions has attracted much attention.2 For example, the group of Yu as well as other research groups have utilized carboxylic acids,3 alcohols,4 amides,5 sulfonamides,6N-methoxy amides,7etc. as weakly coordinating functional groups8 for a series of efficient metal-catalyzed C–H functionalizations (Scheme 1). Beside C–H bond activations, several examples of C–C bond activation with the coordinating moieties as directing groups have also been reported recently.9 However, to the best of our knowledge, the C–C bond activation through a weak-coordination approach has been seldom reported so far. Methylenecyclopropanes (MCPs) and vinylidenecyclopropanes (VDCPs) are both highly strained but readily accessible and adequately reactive molecules which can serve as useful building blocks in organic synthesis.10 They can undergo a variety of ring-opening reactions because the release of cyclopropyl ring strain can provide a thermodynamic driving force for reactions and the π-character of the bonds within the cyclopropane can afford the kinetic opportunity to initiate the unleashing of the strain.11 During our ongoing investigation on the ring-opening reactions of the MCPs and VDCPs in the presence of metal catalysts, we found a new C–C bond activation mode of VDCPs bearing a weakly coordinating group in the presence of Pd(OAc)2 under mild conditions (Scheme 1). Herein, we wish to report the details.
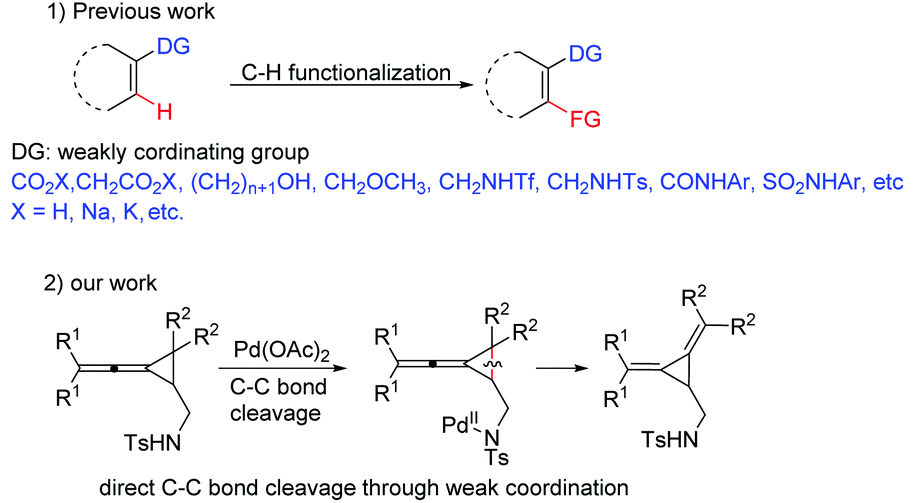 | ||
| Scheme 1 C–H and C–C bond cleavages through weak coordination. | ||
The starting materials sulfonamide-tethered vinylidene-cyclopropanes (VDCPs) 1 were prepared according to the previously reported procedure12 and these functionalized VDCPs were utilized as substrates for further transformation in the presence of Pd catalysts.
The initial examination on the intramolecular rearrangement of VDCPs 1 was carried out upon heating 1a in toluene at 60 °C in the presence of Pd(OAc)2 (10 mol%) and PPh3 (20 mol%) as the ligand under an argon atmosphere. However, complex product mixtures were obtained after 10 h (Table 1, entry 1). In the absence of the PPh3 ligand, the rearranged product 2a was formed in 76% isolated yield (91% NMR yield) (Table 1, entry 2). Its structure was determined by X-ray diffraction and its ORTEP drawing is shown in Fig. 1.13
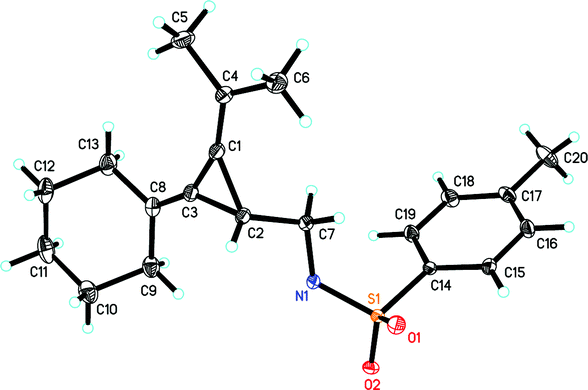 | ||
| Fig. 1 X-ray crystal structure of 2a. | ||
|
|
||||||
|---|---|---|---|---|---|---|
| Entrya | Catalyst | Additive | Solvent | t (h) | T (°C) | Yieldb (%) |
| a The reaction conditions: catalyst (10 mol%), 0.1 M in solvent unless otherwise specified. b The yield was determined by H NMR spectroscopic data using 1,3,5-trimethoxybenzene as an internal standard. c The ligand pph3 (20 mol%) was added. d Isolated yields. e The quaternary ammonium salts were added with 20 mol%. f With catalyst (5 mol%). g With H2O (50 mol%). h Stoichiometric amount of the catalyst was used. i The reactions gave complex product mixtures. | ||||||
| 1 | Pd(OAc)2/PPh3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) c c |
— | Toluene | 5 | 60 | —i |
| 2 | Pd(OAc)2 | — | Toluene | 8 | 60 | 91 (76)d |
| 3 | Pd/C | — | Toluene | 5 | 60 | N. R. |
| 4 | [Pd(η3-C3H5)Cl]2 | — | Toluene | 5 | 60 | —i |
| 5 | Pd(OAc)2(Py)2 | — | Toluene | 5 | 60 | —i |
| 6 | Pd(PhCN)2Cl2 | — | Toluene | 5 | 60 | —i |
| 7 | PdCl2(PPh3)2 | — | Toluene | 5 | 60 | —i |
| 8 | Pd(dppf)(OTf)2 | — | Toluene | 5 | 60 | —i |
| 9 | Pd(dppf)Cl2 | — | Toluene | 5 | 60 | —i |
| 10 | PdCl2 | — | Toluene | 5 | 60 | —i |
| 11 | (2,2′bipy)Pd(OAc)2 | — | Toluene | 5 | 60 | —i |
| 12 | Pd(OAc)2e |
Bu4NCl | Toluene | 10 | 60 | N. R. |
| 13 | Pd(OAc)2e |
PhEt3NCl | Toluene | 10 | 60 | N. R. |
| 14 | Pd(TFA)2 | — | Toluene | 10 | 80 | 30 |
| 15 | AuCl3 | — | Toluene | 10 | 80 | —i |
| 16 | Rh2(OAc)2 | — | Toluene | 10 | 80 | —i |
| 17 | PtCl2 | — | Toluene | 10 | 80 | —i |
| 18 | Cu(OAc)2 | — | Toluene | 10 | 80 | N. R. |
| 19 | Pd(OAc)2 | — | THF | 24 | rt | 70 |
| 20 | Pd(OAc)2 | — | Et2O | 24 | rt | 83 |
| 21 | Pd(OAc)2 | — | CH2Cl2 | 24 | rt | 85 |
| 22 | Pd(OAc)2 | — | DCE | 10 | 60 | 73 |
| 23 | Pd(OAc)2 | — | MeCN | 10 | 60 | 62 |
| 24 | Pd(OAc)2 | — | Toluene | 24 | rt | 85 |
| 25 | Pd(OAc)2 | — | Toluene | 8 | 80 | 91 (76)d |
| 26f | Pd(OAc)2 | — | Toluene | 8 | 80 | 83 |
| 27f | Pd(OAc)2 | — | Toluene | 8 | 100 | 57 |
| 28 | Pd(OAc)2 | O2 | Toluene | 8 | 60 | 45 |
| 29g | Pd(OAc)2 | H2O | Toluene | 8 | 80 | 72 |
| 30h | Pd(OAc)2 | — | Toluene | 8 | 60 | —i |
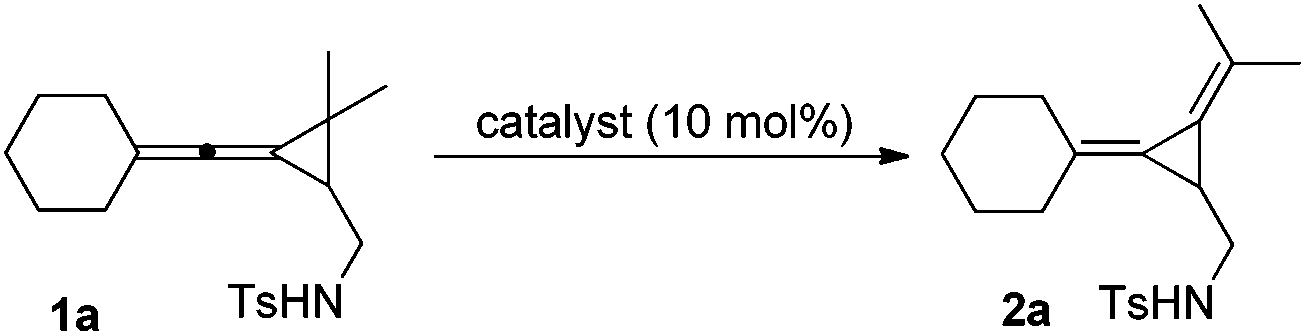
Then we studied other Pd catalysts such as Pd/C, Pd(η3-C3H5)Cl, Pd(OAc)2(Py)2, Pd(PhCN)2Cl2, PdCl2(PPh3)2, Pd(dppf)(OTf)2, Pd(dppf)Cl2, PdCl2 and (2,2′-biPy)Pd(OAc)2 and the results are summarized in Table 1. We found that the reaction almost did not give the desired product cleanly using these Pd catalysts in toluene, suggesting that Pd(OAc)2 is the best Pd catalyst for this reaction (Table 1, entries 3–11). Using Pd(OAc)2 as the catalyst, the addition of Bu4NCl or PhEt3NCl completely stopped the reaction (Table 1, entries 12 and 13). The use of Pd(TFA)2 as the catalyst gave 2a in 30% yield in toluene at 80 °C under otherwise identical conditions (Table 1, entry 14). Other transition metal catalysts such as AuCl3, Rh2(OAc)2 and PtCl2 produced complex product mixtures (Table 1, entries 15–17). No reaction occurred using Cu(OAc)2 (10 mol%) as the catalyst (Table 1, entry 18). The examination of solvent effects revealed that toluene, CH2Cl2 and Et2O were better than THF, DCE (1,2-dichloroethane) and MeCN (Table 1, entries 19–23) since upon heating in DCE (1,2-dichloroethane) or MeCN at 60 °C afforded 2a in 73% or 62% NMR yield, respectively (Table 1, entries 22 and 23). All these results indicated that toluene is best suited to this reaction. The effect of temperature has been also examined in toluene. Upon heating at 80 °C 2a in 91% NMR yield (76% isolated yield) was obtained, which is exactly the same as that at 60 °C (Table 1, entries 2 and 25). Reducing the employed amount of Pd(OAc)2 to 5 mol% produced 2a in 83% yield under otherwise identical conditions (Table 2, entry 26) and increasing the reaction temperature up to 100 °C reduced the yield of 2a to 57% yield in the presence of 5 mol% Pd(OAc)2 (Table 1, entry 27). Furthermore, this reaction should be carried out under an argon atmosphere and anhydrous conditions since under an oxygen atmosphere or in the presence of water also reduced the yield of 2a (Table 1, entries 28 and 29). The use of stoichiometric amount of Pd(OAc)2 gave a complex product mixture (Table 1, entry 30). All these examinations revealed that this reaction should be carried out at 80 °C in toluene in the presence of 10 mol% Pd(OAc)2.
| a Reaction conditions: VDCP 1 (0.2 mmol), Pd(OAc)2 (0.02 mmol), toluene (2 mL). Isolated yields. |
|---|
|
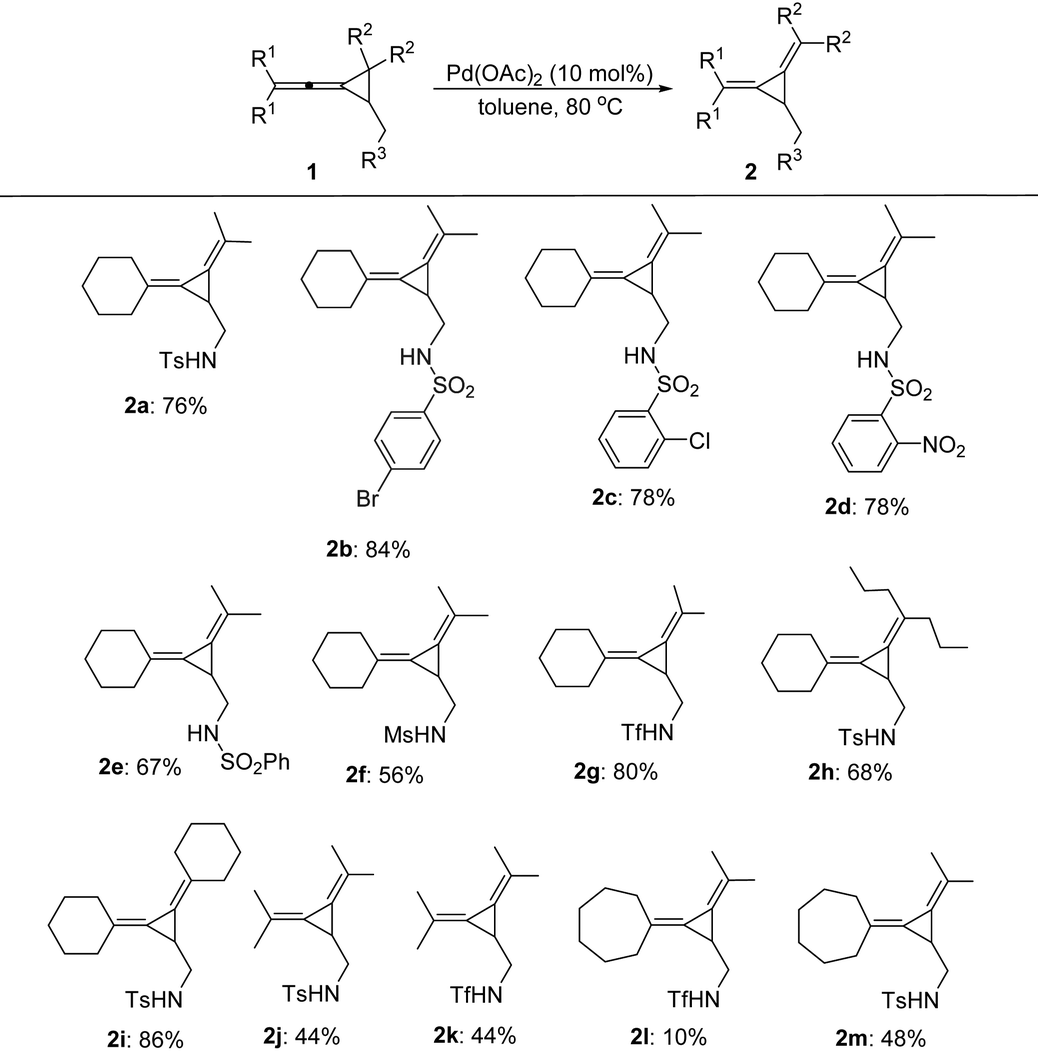
With the identification of the best reaction conditions, we next turned our efforts to investigate the scope and limitations of these intramolecular rearrangement and the results are summarized in Table 2. A variety of VDCPs 1 bearing different sulfonamide substituents have been tested and the corresponding products 2b–2g were obtained in moderate to good yields without the observation of significant electronic effects (Table 2). Substrate 2g having strongly electron-withdrawing trifluoromethylsulfonamide (R3 = triflamide) was also tolerated, affording the corresponding product 2g in 80% yield. A range of different substituents at the cyclopropane or allene moiety of 1 have also been tested, giving the desired products 2h–2m in 10–86% yields. As for substrate 1l having a cycloheptyl substituent (R1 and R1) and a triflamide functional group, the corresponding product 2l was obtained in 10% yield presumably due to the instability of this product at 80 °C.
The control experiment shown in Scheme 2 indicated that when the substituent R3 was a fully substituted sulfonamide group, carbonamides or a free hydroxyl group, none of the desired products could be formed, suggesting that the sulfonamide group (R3) is essential for this reaction.
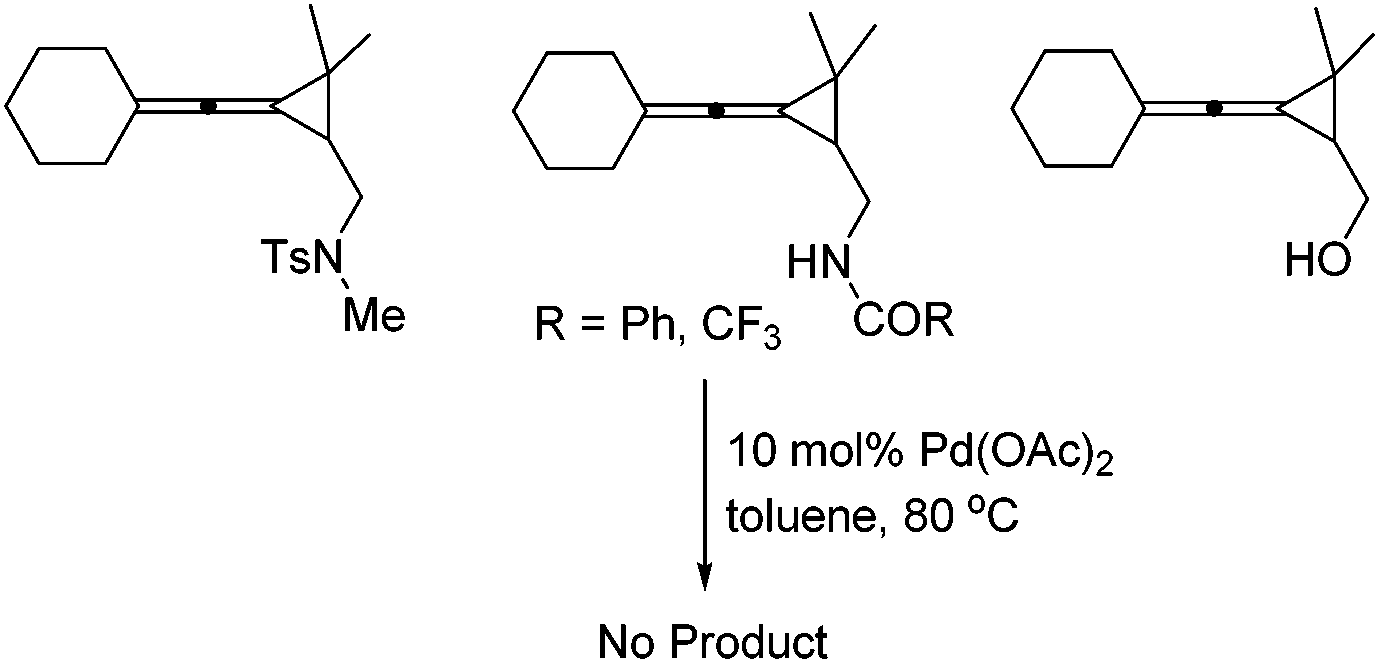 | ||
| Scheme 2 The control experiments. | ||
A plausible reaction mechanism is depicted as below using 1a as a substrate model on the basis of the previous literature and the control experiments (Scheme 3). Since R3 should be a sulfonamide group with a N–H moiety, it may work as a weak-coordinating group for Pd(II) to give intermediate I,5,6 which subsequently undergoes ring-opening upon intramolecular attacking of the NTs moiety to afford the corresponding allylic Pd intermediate II along with the formation of an aziridine. Then, the central carbon of the previous allene moiety attacks the aziridine to afford intermediate III. The protonation of III produces the thermodynamically stable product 2a. The N–H group in sulfonamide is important in proton exchange with Pd(OAc)2 to incorporate the Pd(II) species into the directing group as shown in Scheme 3. The use of the Pd(0) catalyst such as Pd2dba3 and Pd(PPh3)4 as catalysts gave complex product mixtures, rendering that this is a Pd(II) catalyzed process.
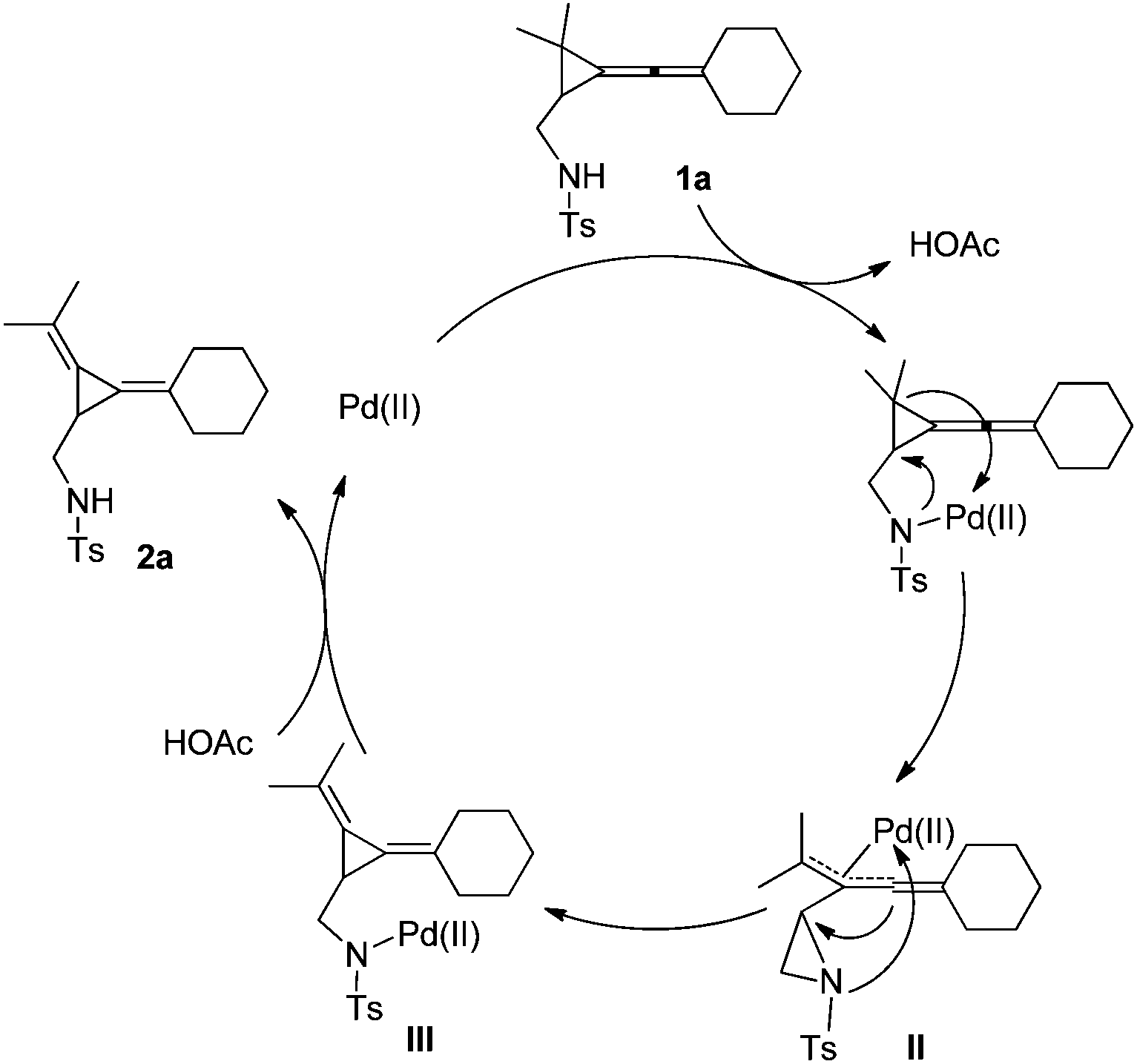 | ||
| Scheme 3 A plausible mechanism for the formation of 2a. | ||
Interestingly, substrate 1q, bearing two phenyl groups at cyclopropane (R2 = phenyl group), produced a complex mixture under the standard conditions. However, upon heating 1q in methanol in the presence of K2CO3 (2 equiv.) afforded another rearranged product 3q in 80% as the E-configuration (Scheme 4). Its structure was determined by X-ray diffraction and its ORTEP drawing is shown in Fig. 2.14 As a comparison, compound 1a was also subjected to the basic conditions, but none of the similar products were formed.
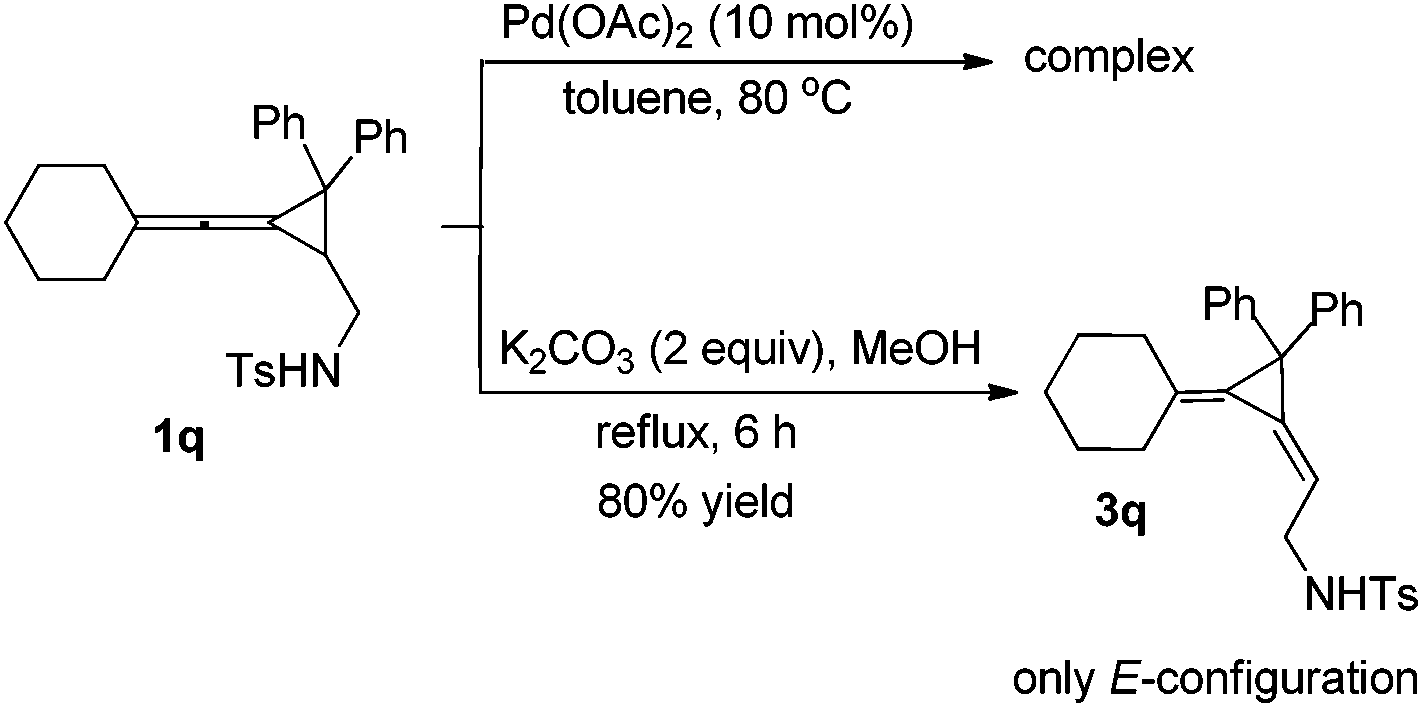 | ||
| Scheme 4 The rearrangement of 1q under basic conditions. | ||
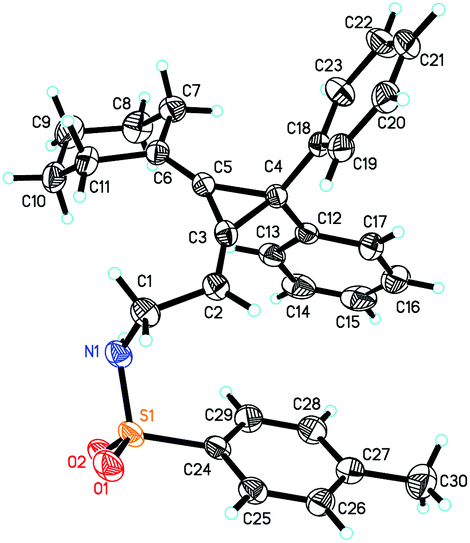 | ||
| Fig. 2 X-ray crystal structure of 3q. | ||
A mechanistic explanation for the intramolecular rearrangement of 1q in the presence of K2CO3 has been proposed in Scheme 5. Firstly, deprotonation of 1q by K2CO3 gives anionic intermediate A, which undergoes intramolecular nucleophilic attack onto the cyclopropane affords anionic aziridine intermediate B accompanied by a ring-opening process. Then, a nucleophilic attack onto the central carbon of allene takes place along with the migration of a double bond, leading to the aziridine ring-opening intermediate C. Protonation of intermediate C gives the corresponding product 3q. The different reaction outcome on phenyl substituted substrate 1q may be due to the stabilization of the anionic intermediate by the phenyl group.
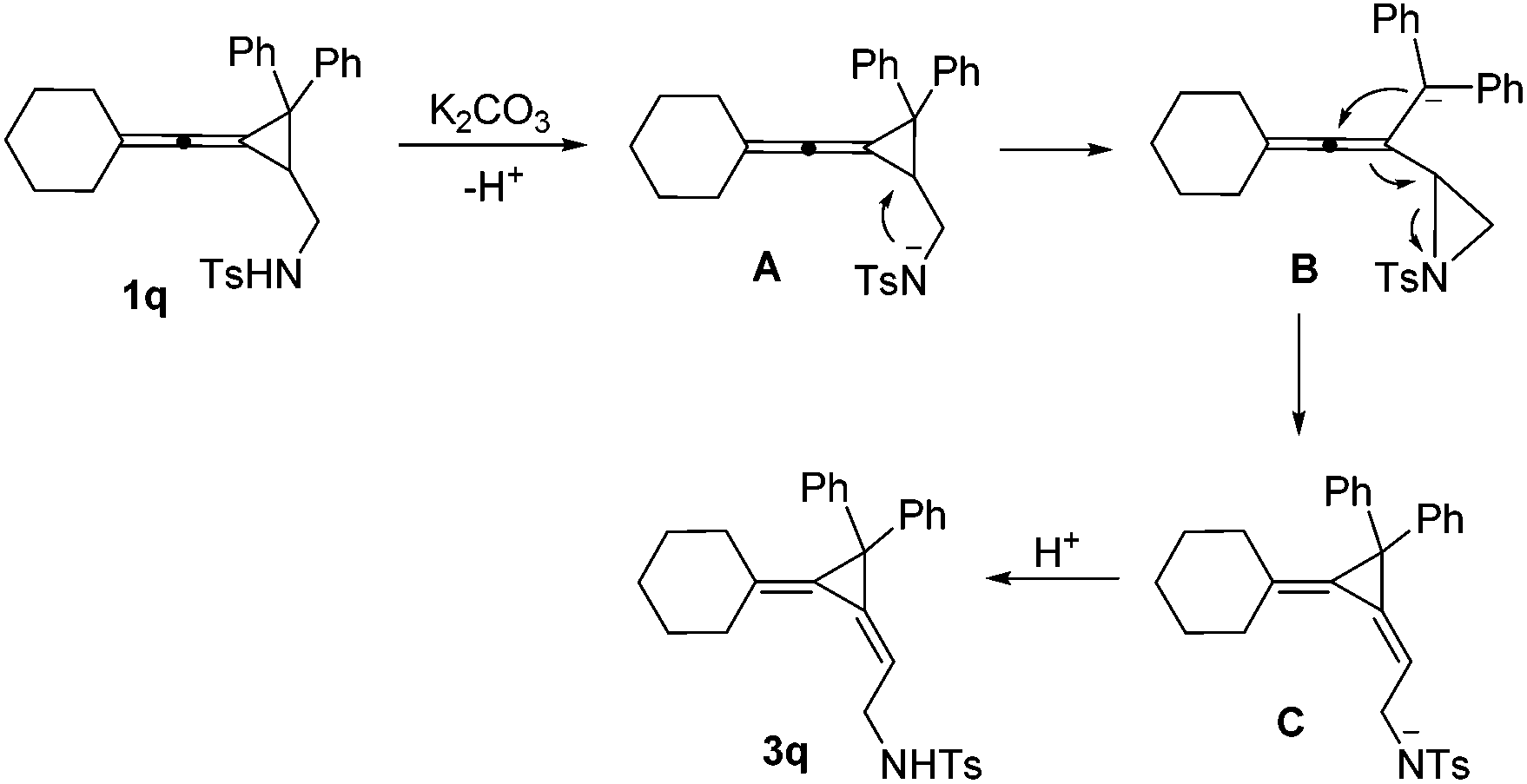 | ||
| Scheme 5 A plausible mechanism for the formation of 3q. | ||
In summary, we have developed a novel C–C bond activation mode of functionalized vinylidenecyclopropanes using a simple and weakly coordinating sulfonamide directing group. Different reaction pathways have been observed when R2 was a phenyl group. The reaction mechanism has also been discussed on the basis of the control experiment and previous results. Further work is underway to elucidate further mechanistic details of these reactions and to understand their scope and limitations in our laboratory.
We are grateful for the financial support from the National Basic Research Program of China (973)-2015CB856603, and the National Natural Science Foundation of China (20472096, 21372241, 21361140350, 20672127, 21421091, 21372250, 21121062, 21302203 and 20732008).
Notes and references
- (a) X.-X. Guo, D.-W. Gu, Z. Wu and W. Zhang, Chem. Rev., 2015, 115, 1622 CrossRef CAS PubMed; (b) H. Huang, X. Ji, W. Wu and H. Jiang, Chem. Soc. Rev., 2015, 44, 1155 RSC; (c) M.-L. Louillat and F. W. Patureau, Chem. Soc. Rev., 2014, 43, 901 RSC; (d) D. G. Musaev, T. M. Figg and A. L. Kaledin, Chem. Soc. Rev., 2014, 43, 5009 RSC; (e) J. Xie, C. Pan, A. Abdukader and C. Zhu, Chem. Soc. Rev., 2014, 43, 5245 RSC; (f) F. Zhang and D. R. Spring, Chem. Soc. Rev., 2014, 43, 6906 RSC; (g) B.-J. Li and Z.-J. Shi, Chem. Soc. Rev., 2012, 41, 5588 RSC; (h) J. Wencel-Delord, T. Droege, F. Liu and F. Glorius, Chem. Soc. Rev., 2011, 40, 4740 RSC; (i) D. Balcells, E. Clot and O. Eisenstein, Chem. Rev., 2010, 110, 749 CrossRef CAS PubMed; (j) E. M. Beck and M. J. Gaunt, Top. Curr. Chem., 2010, 292, 85 CrossRef CAS; (k) D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624 CrossRef CAS PubMed; (l) R. Jazzar, J. Hitce, A. Renaudat, J. Sofack-Kreutzer and O. Baudoin, Chem. – Eur. J., 2010, 16, 2654 CrossRef CAS PubMed; (m) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS PubMed; (n) X. Chen, K. M. Engle, D.-H. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094 CrossRef CAS PubMed; (o) O. Daugulis, H.-Q. Do and D. Shabashov, Acc. Chem. Res., 2009, 42, 1074 CrossRef CAS PubMed; (p) R. Giri, B.-F. Shi, K. M. Engle, N. Maugel and J.-Q. Yu, Chem. Soc. Rev., 2009, 38, 3242 RSC; (q) P. Thansandote and M. Lautens, Chem. – Eur. J., 2009, 15, 5874 CrossRef CAS PubMed; (r) D. Alberico, M. E. Scott and M. Lautens, Chem. Rev., 2007, 107, 174 CrossRef CAS PubMed; (s) E. M. Beccalli, G. Broggini, M. Martinelli and S. Sottocornola, Chem. Rev., 2007, 107, 5318 CrossRef CAS PubMed; (t) L.-C. Campeau, D. R. Stuart and K. Fagnou, Aldrichimica Acta, 2007, 40, 35 CAS.
- (a) L. Ackermann, Acc. Chem. Res., 2014, 47, 281 CrossRef CAS PubMed; (b) S. De Sarkar, W. Liu, S. I. Kozhushkov and L. Ackermann, Adv. Synth. Catal., 2014, 356, 1461 CrossRef CAS PubMed; (c) Y. Rao, Synlett, 2013, 2472 CrossRef CAS PubMed; (d) K. M. Engle, T.-S. Mei, M. Wasa and J.-Q. Yu, Acc. Chem. Res., 2012, 45, 788 CrossRef CAS PubMed; (e) L. Ackermann, Chem. Rev., 2011, 111, 1315 CrossRef CAS PubMed.
- (a) S. Ma, G. Villa, P. S. Thuy-Boun, A. Homs and J.-Q. Yu, Angew. Chem., Int. Ed., 2014, 53, 734 CrossRef CAS PubMed; (b) P. S. Thuy-Boun, G. Villa, D. Dang, P. Richardson, S. Su and J.-Q. Yu, J. Am. Chem. Soc., 2013, 135, 17508 CrossRef CAS PubMed; (c) K. M. Engle, P. S. Thuy-Boun, M. Dang and J.-Q. Yu, J. Am. Chem. Soc., 2011, 133, 18183 CrossRef CAS PubMed.
- (a) Y. Lu, D.-H. Wang, K. M. Engle and J.-Q. Yu, J. Am. Chem. Soc., 2010, 132, 5916 CrossRef CAS PubMed; (b) X. Wang, Y. Lu, H.-X. Dai and J.-Q. Yu, J. Am. Chem. Soc., 2010, 132, 12203 CrossRef CAS PubMed.
- (a) X.-C. Wang, W. Gong, L.-Z. Fang, R.-Y. Zhu, S. Li, K. M. Engle and J.-Q. Yu, Nature, 2015, 519, 334 CrossRef CAS PubMed; (b) G. He, Y. Zhao, S. Zhang, C. Lu and G. Chen, J. Am. Chem. Soc., 2012, 134, 3 CrossRef CAS PubMed; (c) X.-G. Zhang, H.-X. Dai, M. Wasa and J.-Q. Yu, J. Am. Chem. Soc., 2012, 134, 11948 CrossRef CAS PubMed.
- (a) C. Feng, D. Feng and T.-P. Loh, Chem. Commun., 2015, 51, 342 RSC; (b) C. Feng, D. Feng, Y. Luo and T.-P. Loh, Org. Lett., 2014, 16, 5956 CrossRef CAS PubMed; (c) K. S. L. Chan, M. Wasa, X. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2011, 50, 9081 CrossRef CAS PubMed; (d) H.-X. Dai, A. F. Stepan, M. S. Plummer, Y.-H. Zhang and J.-Q. Yu, J. Am. Chem. Soc., 2011, 133, 7222 CrossRef CAS PubMed.
- (a) Y.-J. Liu, H. Xu, W.-J. Kong, M. Shang, H.-X. Dai and J.-Q. Yu, Nature, 2014, 515, 389 CrossRef CAS PubMed; (b) D.-H. Wang, M. Wasa, R. Giri and J.-Q. Yu, J. Am. Chem. Soc., 2008, 130, 7190 CrossRef CAS PubMed.
- (a) B. E. Haines and D. G. Musaev, ACS Catal., 2015, 5, 830 CrossRef CAS; (b) G. Li, L. Wan, G. Zhang, D. Leow, J. Spangler and J.-Q. Yu, J. Am. Chem. Soc., 2015, 137, 4397 Search PubMed; (c) W. Yang, S. Ye, D. Fanning, T. Coon, Y. Schmidt, P. Krenitsky, D. Stamos and J.-Q. Yu, Angew. Chem., Int. Ed., 2015, 54, 2501 CrossRef CAS PubMed; (d) J. Kim and S. Chang, Angew. Chem., Int. Ed., 2014, 53, 2203 CrossRef CAS PubMed; (e) S. Li, G. Chen, C.-G. Feng, W. Gong and J.-Q. Yu, J. Am. Chem. Soc., 2014, 136, 5267 CrossRef CAS PubMed; (f) X. Sun, G. Shan, Y. Sun and Y. Rao, Angew. Chem., Int. Ed., 2013, 52, 4440 CrossRef CAS PubMed; (g) X.-C. Wang, Y. Hu, S. Bonacorsi, Y. Hong, R. Burrell and J.-Q. Yu, J. Am. Chem. Soc., 2013, 135, 10326 CrossRef CAS PubMed; (h) Z. Ren, F. Mo and G. Dong, J. Am. Chem. Soc., 2012, 134, 16991 CrossRef CAS PubMed; (i) P. E. Zhichkin, S. G. Krasutsky and R. Krishnamoorthy, Synlett, 2010, 3039 CrossRef CAS PubMed.
- (a) R. Zeng and G. Dong, J. Am. Chem. Soc., 2015, 137, 1408 CrossRef CAS PubMed; (b) A. Dermenci, J. W. Coe and G. Dong, Org. Chem. Front., 2014, 1, 567 RSC; (c) A. Korotvicka, D. Necas and M. Kotora, Curr. Org. Chem., 2012, 16, 1170 CrossRef CAS; (d) K. Ruhland, Eur. J. Org. Chem., 2012, 2683 CrossRef CAS PubMed; (e) M. Murakami and T. Matsuda, Chem. Commun., 2011, 47, 1100 RSC; (f) T. Seiser, T. Saget, D. N. Tran and N. Cramer, Angew. Chem., Int. Ed., 2011, 50, 7740 CrossRef CAS PubMed; (g) C.-H. Jun and J.-W. Park, Top. Organomet. Chem., 2007, 24, 117 CrossRef CAS; (h) D. Necas and M. Kotora, Curr. Org. Chem., 2007, 11, 1566 CrossRef CAS; (i) T. Kondo and T.-a. Mitsudo, Chem. Lett., 2005, 34, 1462 CrossRef CAS; (j) C.-H. Jun, Chem. Soc. Rev., 2004, 33, 610 RSC; (k) M. E. van der Boom and D. Milstein, Chem. Rev., 2003, 103, 1759 CrossRef CAS PubMed; (l) C. Perthuisot, B. L. Edelbach, D. L. Zubris, N. Simhai, C. N. Iverson, C. Muller, T. Satoh and W. D. Jones, J. Mol. Catal. A: Chem., 2002, 189, 157 CrossRef CAS; (m) L. Giannini, S. Dovesi, E. Solari, C. Floriani, A. Chiesi-Villa and C. Rizzoli, Angew. Chem., Int. Ed., 1999, 38, 807 CrossRef CAS; (n) M. Murakami and Y. Ito, Top. Organomet. Chem., 1999, 3, 97 CrossRef CAS; (o) M. Gozin, A. Weisman, Y. Ben-David and D. Milstein, Nature, 1993, 364, 699 CrossRef CAS PubMed; (p) R. H. Crabtree, Chem. Rev., 1985, 85, 245 CrossRef CAS.
- (a) A. Brandi, S. Cicchi, F. M. Cordero and A. Goti, Chem. Rev., 2014, 114, 7317 CrossRef CAS PubMed; (b) D.-H. Zhang, X.-Y. Tang and M. Shi, Acc. Chem. Res., 2014, 47, 913 CrossRef CAS PubMed; (c) M. Shi, J.-M. Lu, Y. Wei and L.-X. Shao, Acc. Chem. Res., 2012, 45, 641 CrossRef CAS PubMed; (d) Y. Yamamoto, Chem. Rev., 2012, 112, 4736 CrossRef CAS PubMed; (e) L. Yu and R. Guo, Org. Prep. Proced. Int., 2011, 43, 209 CrossRef CAS PubMed; (f) A. Masarwa and I. Marek, Chem. – Eur. J., 2010, 16, 9712 CrossRef CAS PubMed; (g) M. Shi, L.-X. Shao, J.-M. Lu, Y. Wei, K. Mizuno and H. Maeda, Chem. Rev., 2010, 110, 5883 CrossRef CAS PubMed; (h) M. Rubin, M. Rubina and V. Gevorgyan, Chem. Rev., 2007, 107, 3117 CrossRef CAS PubMed; (i) I. Nakamura and Y. Yamamoto, Adv. Synth. Catal., 2002, 344, 111 CrossRef CAS.
- (a) B. Mueller, T. Bally, R. Pappas and F. Williams, J. Am. Chem. Soc., 2010, 132, 14649 CrossRef CAS PubMed; (b) A. De Meijere and S. Blechert, NATO Advanced Science Institutes Series, Series C Mathematical and Physical Sciences, Vol. 273: Strain and Its Implications in Organic Chemistry, Kluwer Academic Publ., 1989 Search PubMed; (c) A. De Meijere, Top. Curr. Chem., 1988, 144, 217 Search PubMed.
- (a) B.-L. Lu, Y. Wei and M. Shi, Organometallics, 2012, 31, 4601 CrossRef CAS; (b) B.-L. Lu and M. Shi, Eur. J. Org. Chem., 2011, 243 CrossRef CAS PubMed; (c) B.-L. Lu and M. Shi, Angew. Chem., Int. Ed., 2011, 50, 12027 CrossRef CAS PubMed; (d) B.-L. Lu, Y. Wei and M. Shi, Chem. – Eur. J., 2010, 16, 10975 CrossRef CAS PubMed.
- The CIF data of 2a have been deposited in CCDC 958985 (see ESI†).
- The CIF data of 3q have been deposited in CCDC 1031145 (see ESI†).
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data of new compounds. CCDC 958985 and 1031145. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5qo00127g |
| This journal is © the Partner Organisations 2015 |