
Palladium-catalyzed direct ortho C–O bond construction of azoxybenzenes with carboxylic acids and alcohols†
Duo
Zhang
a,
Xiuling
Cui
*ab,
Fangfang
Yang
a,
Qianqian
Zhang
a,
Yu
Zhu
a and
Yangjie
Wu
*a
aDepartment of Chemistry, Henan Key Laboratory of Chemical Biology and Organic Chemistry, Key Laboratory of Applied Chemistry of Henan Universities, Zhengzhou University, Zhengzhou 450052, P. R. China. E-mail: cuixl@zzu.edu.cn
bXiamen Key Laboratory of Ocean and Gene Drugs, School of Biomedical Sciences, Institute of Molecular Medicine of Huaqiao University and Engineering Research Centre of Molecular Medicine of the Chinese Education Ministry, Xiamen, Fujian 361021, P. R. China
First published on 3rd June 2015
Abstract
A Pd(II) catalyzed C–O bond formation of azoxybenzenes has been developed. An N-atom served as a directing group, as confirmed by the single crystal X-ray diffraction analysis of the palladium complex. The products were obtained in moderate to excellent yields for 29 examples. This protocol features high regioselectivity, wide functional group tolerance and atom economy.
Introduction
Azoxybenzenes are widely applied in pharmaceuticals,1a oxidants,1b–d photochemistry,1e spectroscopy,1f,g polymer inhibitors and stabilizers.1h In light of their importance, the development of azoxybenzene derivatives has been a hot topic. Nowadays direct C–H bond functionalization has been established to build various azoxybenzene derivatives. The transition-metal-catalyzed ortho-halogenation,2ortho-acylation,3ortho-arylation,4ortho-alkoxylation,5ortho-acyloxyation,6ortho-amidation,7ortho-sulfonylation,8ortho-phosphorylation,9ortho-nitration10 and cyclization11 of azobenzenes have been developed rapidly. Compared with the formation of C–C, C–S and C–N bonds, the reports on the C–O bond formation based metal-catalyzed C–H bonds activation are less well established, maybe due to the electronegativities of the oxygen atom as well as its strong coordination with metals.5a,12 To the best of our knowledge, there are only three examples of building the C–O bond of azobenzenes in the literature as shown in Scheme 1. Acyloxyation of the azobenzene was reported by the Sanford6a and Cárdenas groups6bvia palladium-catalyzed C–H acyloxyation of azobenzene with PhI(OAc)2 and Zeng6d obtained the acyloxylated azobenzene via palladium-catalyzed acyloxyation of azobenzene with aryl acylperoxides. Sun5a presented alkoxylation of the azobenzene via palladium-catalyzed alkoxylation of azobenzene with methanol. Additionally, the previous studies by Wang,13 Zang14 and our group15 indicated that the introduction of an azoxy group into the azobenzenes can not only bring about a high regioselectivity, but also enhance the reactivity of the target C–H bond. In continuation of our effort on C–H functionalization of azoxybenzenes, we embark on the development of a simple and efficient procedure for various acyloxyazoxybenzenes and alkoxylazoxybenzenes based on palladium-catalyzed direct C–H bond activation with high efficiency and regioselectivity under mild conditions. Compared to azobenzenes, azoxybenzenes exhibited higher regioselectivity and resulted in milder reaction conditions.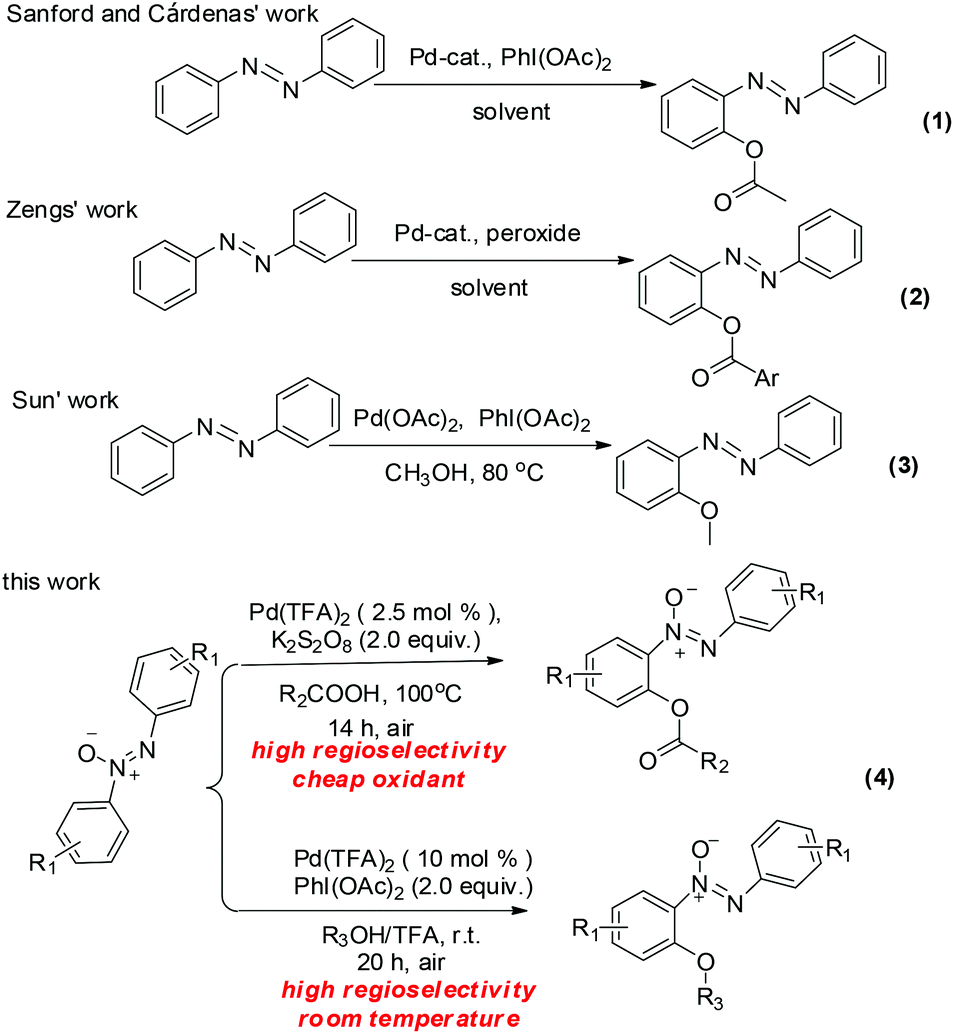 | ||
| Scheme 1 Palladium-catalyzed direct C–O bond construction of diazobenzenes. | ||
Results and discussion
Initially, the cross-coupling of azoxybenzene 1a and acetic acid 2a was chosen as a model reaction to screen various reaction parameters. The results are summarized in Table 1. When the reaction was carried out in carboxylic acids at 100 °C with 10 mol% Pd(OAc)2 as a catalyst and K2S2O8 (2.0 equiv.) as an oxidant, the desired o-acyloxyazoxybenzene 3a was afforded in 79% yield (entry 1, Table 1). Subsequently, a further screening of Pd(dba)2, PdCl2, and Pd(TFA)2 salts indicated that Pd(TFA)2 was the best catalyst and could provide 3a in 86% yield (entries 1–4, Table 1). Other oxidants, such as TBHP, DTBP, Ag2CO3, AgNO3, Ag2O and AgOAc, turned out to be inferior to K2S2O8 (entries 5–10, Table 1). When prolonging the reaction time to 10 hours, the yield was not improved significantly (entry 11, Table 1). The catalyst loading was reduced to 2.5 mol% and the reaction time was 14 hours, the product 3a was obtained in 86% yield (entry 12, Table 1). After surveying the reaction parameters, the optimal reaction conditions were determined as follows: azoxybenzene (0.2 mmol), Pd(TFA)2 (2.5 mol%), and K2S2O8 (2.0 equiv.) in acetic acid (2.0 ml) in a 100 °C oil bath under air for 14 hours.|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Oxidant | Time | Yieldb % |
| a Reaction conditions: azoxybenzene (1a) (0.2 mmol), acetic acid (2a) (2.0 ml), catalyst (10 mol%), oxidant (2.0 equiv.), under air, 100 °C. b Isolated yield based on 1a. c Pd(TFA)2 (2.5 mol%). | ||||
| 1 | Pd(OAc)2 | K2S2O8 | 6 h | 79 |
| 2 | Pd(dba)2 | K2S2O8 | 6 h | 16 |
| 3 | Pd(Cl)2 | K2S2O8 | 6 h | 7 |
| 4 | Pd(TFA)2 | K2S2O8 | 6 h | 86 |
| 5 | Pd(TFA)2 | TBHP | 6 h | nr |
| 6 | Pd(TFA)2 | DTBP | 6 h | nr |
| 7 | Pd(TFA)2 | AgNO3 | 6 h | Trace |
| 8 | Pd(TFA)2 | Ag2CO3 | 6 h | nr |
| 9 | Pd(TFA)2 | Ag2O | 6 h | nr |
| 10 | Pd(TFA)2 | AgOAc | 6 h | nr |
| 11 | Pd(TFA)2 | K2S2O8 | 10 h | 89 |
| 12c | Pd(TFA)2 | K2S2O8 | 14 h | 86 |
The alkoxylation of azoxybenzene with alcohols was investigated. By changing the coupling partner to methanol, methoxyazoxybenzene was achieved in 22% yield (entry 1, Table 2). When the loading of Pd(TFA)2 was increased to 10 mol%, product 4a was obtained in 34% yield (entry 2, Table 2). Inspired by the previous studies, suitable additives might bring crucial effects on the C–H bond activation.16 When 1.2 mmol (6.0 equiv.) of CF3COOH were added, the yield was improved to 50% (entry 3, Table 2). Compared to K2S2O8, PhI(OAc)2 was better (entry 3 vs. entry 4, Table 2). To our surprise, when the reaction occurred at room temperature, the yield was not significantly changed (entry 5, Table 2). When the TFA loading was increased to 12.0 equiv. and 18.0 equiv., product 4a was obtained in 73% and 80% yields, respectively (entries 6 and 7, Table 2). So, the optimal reaction conditions were determined: azoxybenzene (0.2 mmol), Pd(TFA)2 (10 mol%), K2S2O8 (2.0 equiv.), and TFA (18.0 equiv.) in methanol (2.0 ml) at room temperature under air for 20 hours.
|
|
||||
|---|---|---|---|---|
| Entry | Oxidant | Additive | T | Yieldb % |
| a Reaction conditions: azoxybenzene (1a) (0.2 mmol), MeOH (2a′) (2.0 ml), Pd(TFA)2 (10 mol%), oxidant (2.0 equiv.), under air, 20 h. b Isolated yield based on 1a. c Pd(TFA)2 (2.5 mol%). d Room temperature. | ||||
| 1c | K2S2O8 | None | 100 | 22 |
| 2 | K2S2O8 | None | 100 | 34 |
| 3 | K2S2O8 | TFA (6.0 equiv.) | 100 | 50 |
| 4 | PhI(OAc)2 | TFA (6.0 equiv.) | 100 | 57 |
| 5d | PhI(OAc)2 | TFA (6.0 equiv.) | R.T. | 58 |
| 6d | PhI(OAc)2 | TFA (12.0 equiv.) | R.T. | 73 |
| 7d | PhI(OAc)2 | TFA (18.0 equiv.) | R.T. | 80 |
With the optimized reaction conditions in hand, the scope of the reaction between azoxybenzene and carboxylic acids was assessed under the optimized conditions (Table 1). A variety of linear carboxylic acids could be coupled with azoxybenzene (1a), affording the ortho-acyloxyation of azoxybenzenes in 47–86% yields (3a–3e, Scheme 2). The carboxylic acids with α or β steric hindrances were suitable substrates. For example, isovaleric acid, isobutyric acid and trimethylacetic acid gave the corresponding products in 65%, 62% and 44% yields, respectively (3f–3h, Scheme 2). Next, we explored the azoxybenzenes substrates, and found that the procedure can be widely applied. The 2,2′-dimethyl could promote this reaction and give the desired products in 97% yield (3i, Scheme 2). While the 3,3′-dimethyl azoxybenzene provided the product in 65% yield and 3,3′-dibromo azoxybenzene provided the product in 30% yield (3j–3k, Scheme 2). The successful conversion of 4,4′-difluoro, 4,4′-dichloro, 4,4′-ditrifluoromethoxy and 4,4′-diisopropyl azoxybenzenes showed that electron-withdrawing and electron-donating groups substituted in substrates are tolerated (3l–3o, Scheme 2). It is worthy of note that these standard reaction conditions were applied to the unsymmetrical azoxybenzene, affording the corresponding product with only one isomer in 77% yield (3p, Scheme 2). When the 4,4′-dimethyl azoxybenzene reacted with propionic acid, 38% yield was obtained (3q, Scheme 2). The structure of 3q was confirmed by single crystal X-ray diffraction and shown in Fig. 1 in the ESI.†
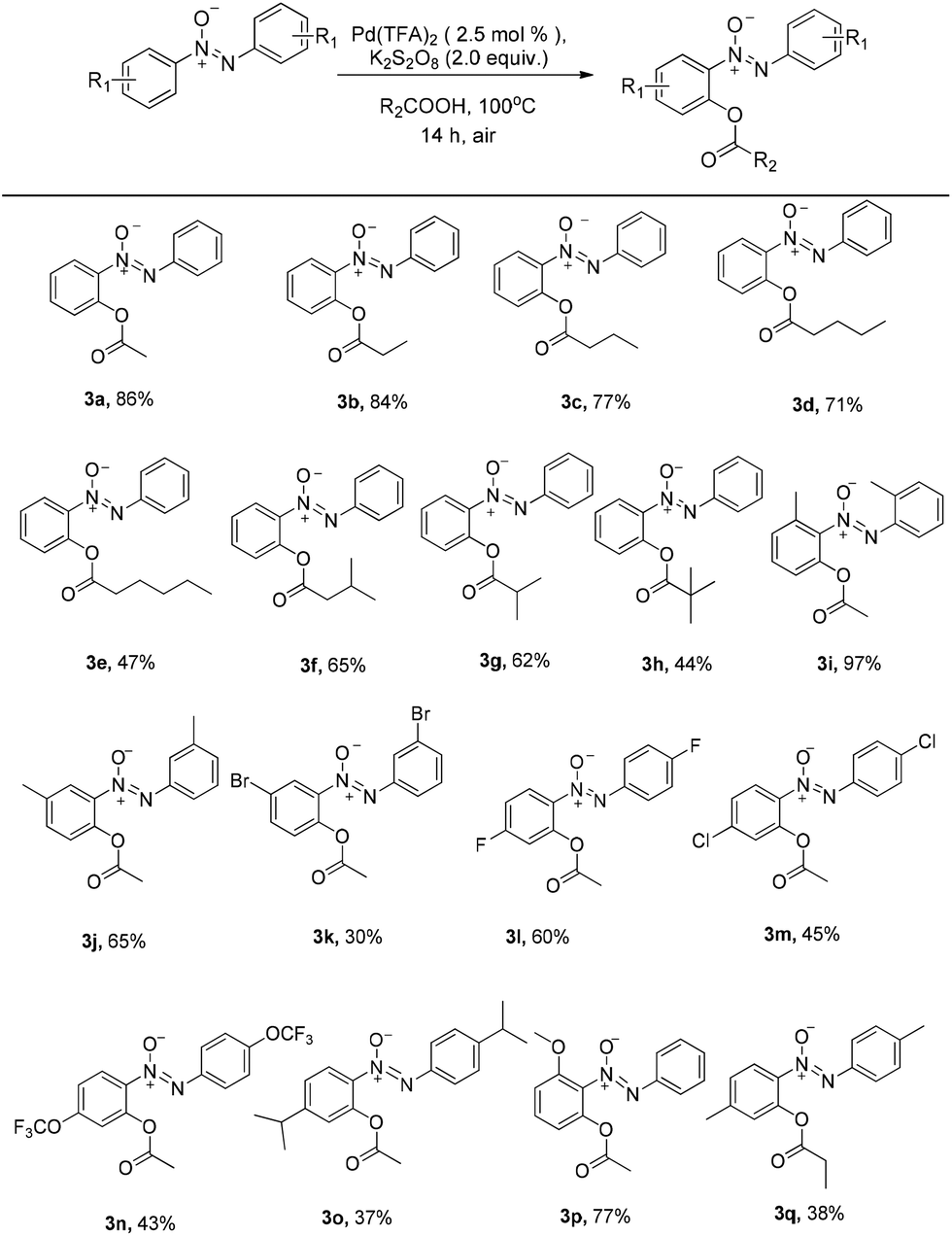 | ||
| Scheme 2 The scope of palladium-catalyzed ortho C–O formation of azoxybenzenes with carboxylic acids. Reaction conditions: azoxybenzenes (0.2 mmol), Pd(TFA)2 (2.5 mol%), K2S2O8 (2.0 equiv.), carboxylic acids (2.0 ml), 100 °C, under air for 14 hours. Isolated yield. | ||
A similar rule applies to azoxybenzene (1a) coupled with alcohols. A variety of linear or branched alcohols could react with azoxybenzenes (1a) to give products 4a–4g. Linear alcohols, such as methanol, ethanol, propanol, butanol and hexylalcohol could be coupled well with azoxybenzene (1a), affording products in 41–80% yields (4a–4e, Scheme 3). The branched alcohols with the steric hindrance of isobutanol and isopropanol did not significantly affect this transformation, providing the desired products in 38% and 34% yields (4f–4g, Scheme 3). Moreover, the steric effect was observed when methyl-substituted azoxybenzenes at their ortho-, meta- and para-positions were also suitable substrates and successfully afforded the desired products in 94%, 81% and 54% yields, respectively (4h–4j, Scheme 3). The more electron-donating the groups on the para-positions, the lower the yield obtained (4jvs.4kScheme 3). As an unsymmetrical substrate, 2-OCH3 azoxybenzene also proceeded well with excellent regioselectivity and gave the product in 79% yield (4lScheme 3). The structure of 4l was confirmed by single crystal X-ray diffraction and shown in Fig. 1 in the ESI.†
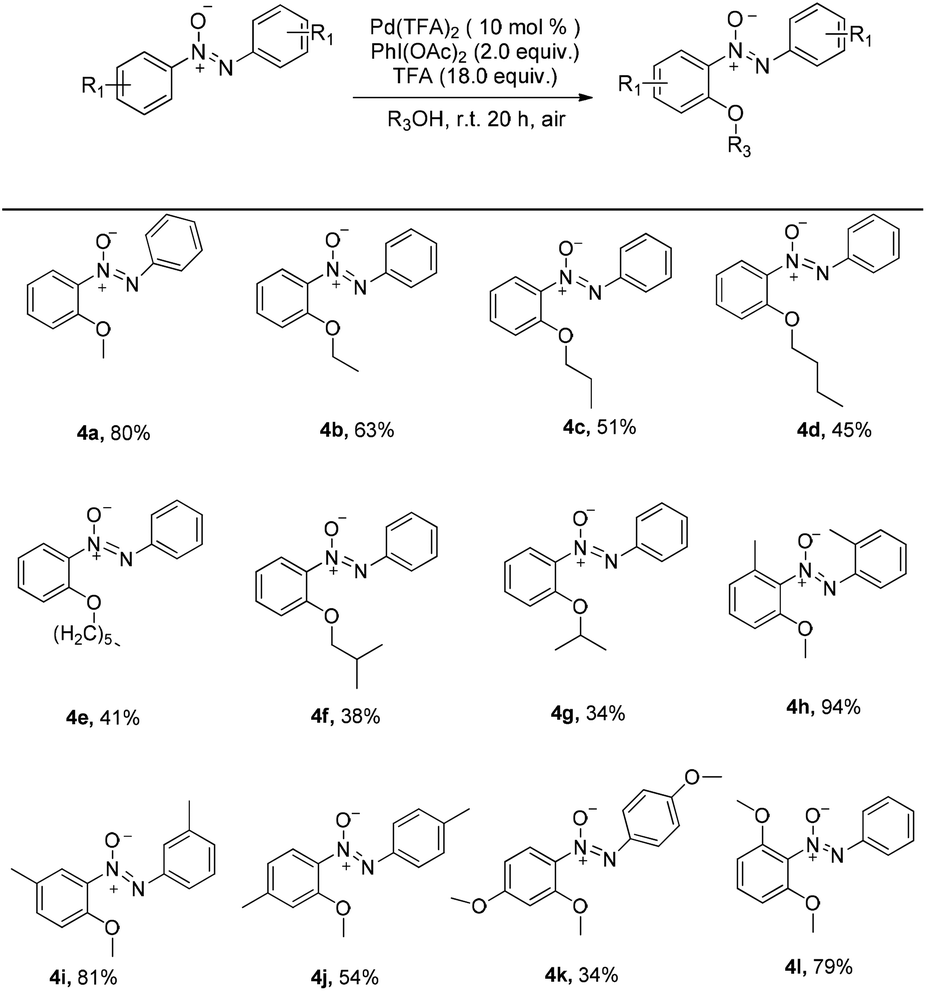 | ||
| Scheme 3 The scope of palladium catalyzed ortho C–O formation of azoxybenzenes with alcohols. Reaction conditions: azoxybenzenes (0.2 mmol), alcohols (2.0 ml), Pd(TFA)2 (10 mol%), PhI(OAc)2 (2.0 equiv.), TFA (18.0 equiv.), room temperature, under air for 20 h. Isolated yield. | ||
To clarify the reaction mechanism, 4.0 equiv. of TEMPO was added to the reactions under the optimized conditions, respectively and no desirable products were detected. Therefore, it suggested that these reactions involve a free radical pathway (Scheme 4). Although the details of the mechanism of this C–O formation remain to be elucidated, a possible mechanism is outlined based on the earlier literature and our preliminary studies.17 The nitrogen atom in azoxybenzene 1a firstly reacted with Pd(TFA)2 to form palladacycle complex I (Fig. 1) through the ortho-C–H bond activation, which accounted for the high regioselectivity in the reactions. Next, the palladium(II) intermediate was reacted with the alkoxyl radical18 or carbonyl oxygen radicals6d,19 to form a palladium(III) or palladium(IV) complex II.20 The reductive elimination of complex II gave the C–O coupled product accompanied by the regeneration of the palladium(II) catalyst.
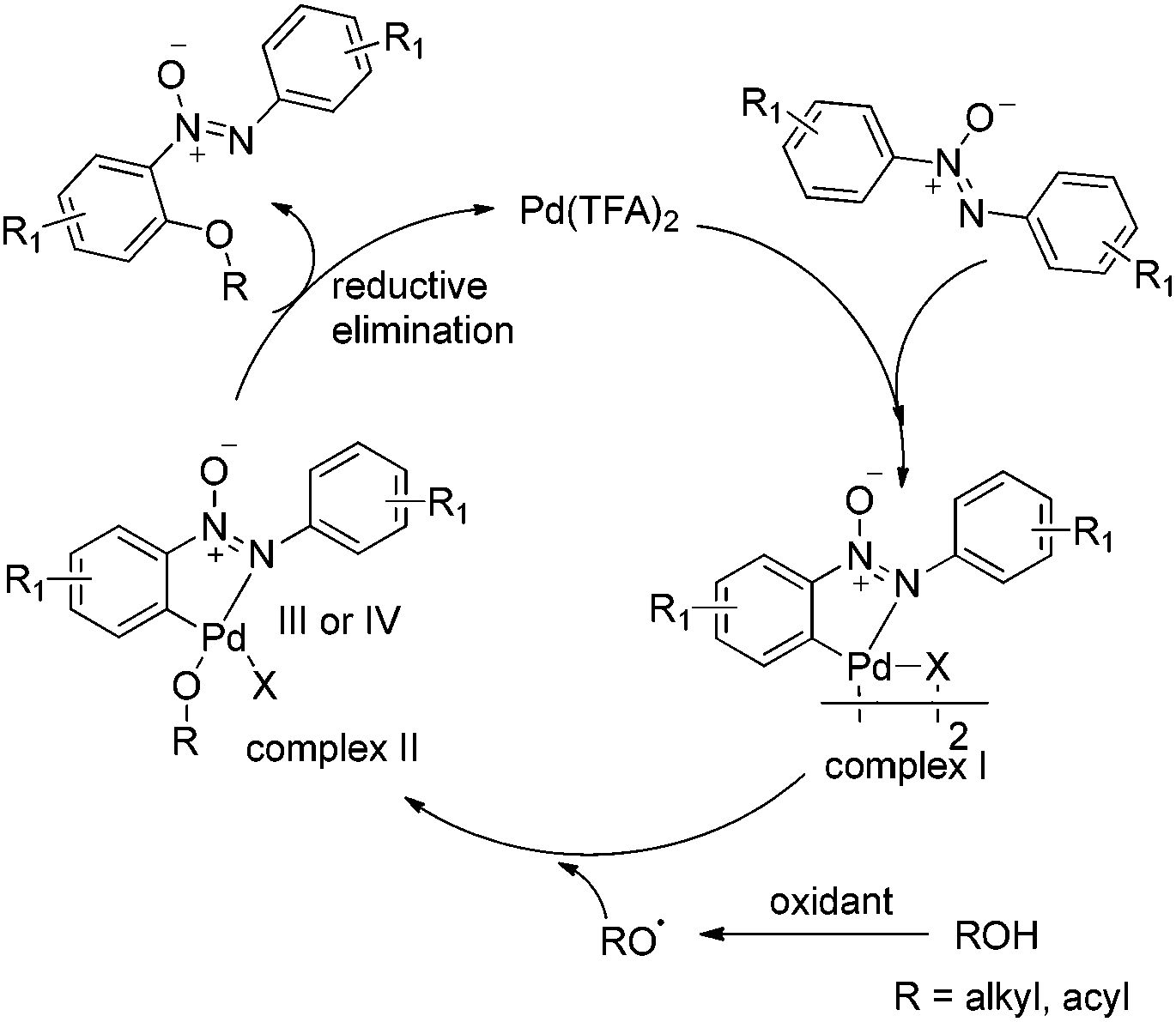 | ||
| Scheme 4 Proposed reaction mechanism. | ||
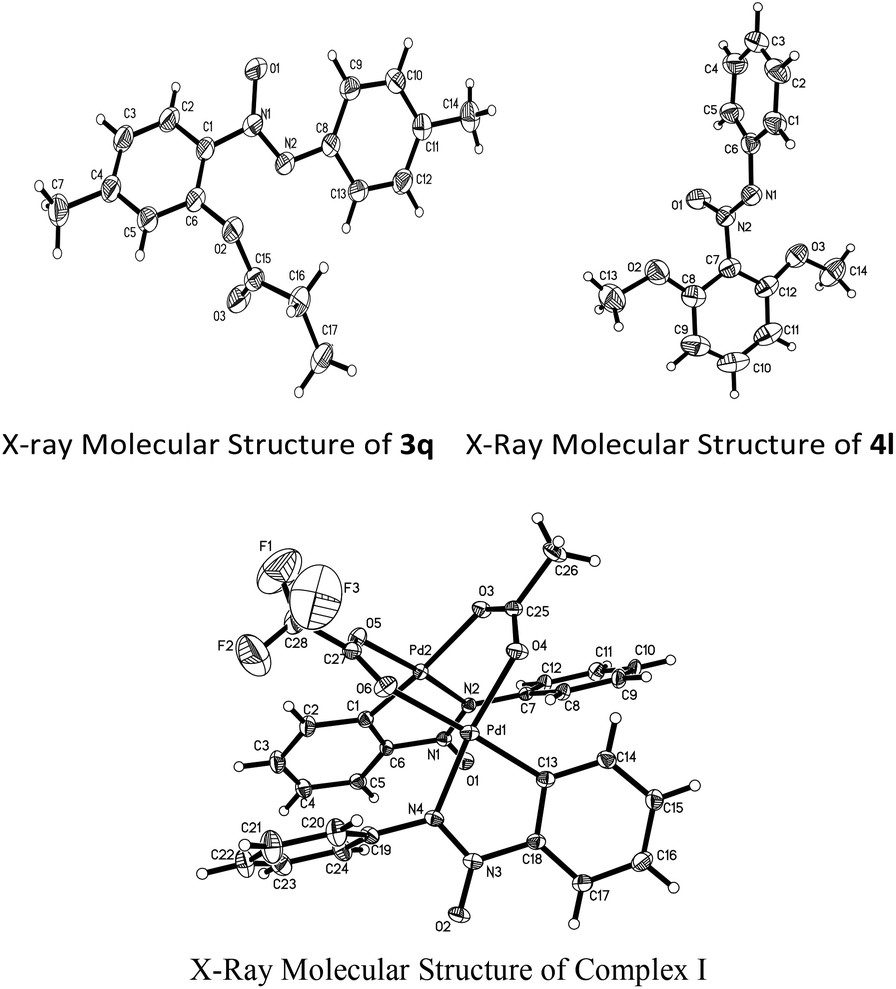 | ||
| Fig. 1 The X-ray molecular structures of 3q, 4l and complex I. | ||
Conclusions
In summary, we have developed a Pd(II) catalyzed C–O bond formation of azoxybenzenes with carboxylic acids and alcohols. An N-atom served as a directing group, as confirmed by single crystal X-ray diffraction analysis. Both acyloxyazoxybenzenes and alkoxylazoxybenzenes were obtained smoothly by these processes. This protocol features high regioselectivity, wide functional group tolerance and atom economy. Further investigations to expand the substrate scope and application of such chemistry in organic synthesis are underway.Acknowledgements
We gratefully acknowledge the National Natural Science Foundation of China (21172200) and Zhengzhou University.Notes and references
- For applications of azoxybenzenes, see: (a) H. Takahashi, T. Ishioka, Y. Koiso, M. Sodeoka and Y. Hashimoto, Biol. Pharm. Bull., 2000, 23, 1387 CrossRef CAS; (b) V. M. Dziomko and K. A. Dunaevskaya, Zh. Obshch. Khim., 1961, 31, 3385 CAS; (c) L. Giuseppe and R. Amerigo, Ann. Chim., 1956, 46, 296 Search PubMed; (d) A. F. Worth and L. Kenneth, Trans. Kans. Acad. Sci., 1932, 35, 146 CrossRef; (e) M. Ōkubo, H. Hyakutake and N. Taniguchi, Bull. Chem. Soc. Jpn., 1988, 61, 3005 CrossRef; (f) A. Barth, J. E. T. Corrie, M. J. Gradwell, Y. Maeda, W. Mäntele, T. Meier and D. R. Trentham, J. Am. Chem. Soc., 1997, 119, 4149 CrossRef CAS; (g) M. A. Berwick and R. E. Rondeau, J. Org. Chem., 1972, 37, 2409 CrossRef CAS; (h) J. M. Huang, J. F. Kuo and C. Y. Chen, J. Appl. Polym. Sci., 1995, 55, 1217 CrossRef CAS PubMed.
- X. T. Ma and S. K. Tian, Adv. Synth. Catal., 2013, 355, 337 CAS.
- (a) H. Y. Song, D. Chen, C. Pi, X. L. Cui and Y. J. Wu, J. Org. Chem., 2014, 79, 2955 CrossRef CAS PubMed; (b) H. J. Li, P. H. Li, Q. Zhao and L. Wang, Chem. Commun., 2013, 49, 9170 RSC; (c) Z. Y. Li, D. D. Li and G. W. Wang, J. Org. Chem., 2013, 78, 10414 CrossRef CAS PubMed; (d) F. Xiong, C. Qian, D. E. Lin, W. Zeng and X. X. Lu, Org. Lett., 2013, 15, 5444 CrossRef CAS PubMed; (e) H. J. Li, P. H. Li and L. Wang, Org. Lett., 2013, 15, 620 CrossRef CAS PubMed; (f) H. Tang, C. Qian, D. E. Lin, H. F. Jiang and W. Zeng, Adv. Synth. Catal., 2014, 356, 519 CrossRef CAS PubMed.
- S. Miyamura, H. Tsurugi, T. Satoh and M. Miura, J. Organomet. Chem., 2008, 693, 2438 CrossRef CAS PubMed.
- (a) Z. W. Yin, X. Q. Jiang and P. P. Sun, J. Org. Chem., 2013, 78, 10002 CrossRef CAS PubMed; (b) S. Shi and C. Kuang, J. Org. Chem., 2014, 79, 6105 CrossRef CAS PubMed.
- (a) A. R. Dick, K. L. Hull and M. S. Sanford, J. Am. Chem. Soc., 2004, 126, 2300 CrossRef CAS PubMed; (b) F. Tato, A. García-Domínguez and D. J. Cárdenas, Organometallics, 2013, 32, 7487 CrossRef CAS; (c) B. C. Kocher, C. Weder and P. Smith, Adv. Funct. Mater., 2003, 13, 427 CrossRef PubMed; (d) C. Qian, D. G. Lin, Y. F. Deng, X. Q. Zhang, H. F. Jiang, G. Miao, X. H. Tang and W. Zeng, Org. Biomol. Chem., 2014, 12, 5866 RSC.
- X. F. Jia and J. Han, J. Org. Chem., 2014, 79, 4180 CrossRef CAS PubMed.
- D. Zhang, X. L. Cui, Q. Q. Zhang and Y. J. Wu, J. Org. Chem., 2015, 80, 1517 CrossRef CAS PubMed.
- G. Hong, D. Mao, S. Y. Wu and L. M. Wang, J. Org. Chem., 2014, 79, 10629 CrossRef CAS PubMed.
- J. W. Dong, B. Jin and P. P. Sun, Org. Lett., 2014, 16, 4540 CrossRef CAS PubMed.
- (a) K. Takagi, M. Al-Amin, N. Hoshiya, J. Wouters, H. Sugimoto, Y. Shiro, H. Fukuda, S. Shuto and M. Arisawa, J. Org. Chem., 2014, 79, 6366 CrossRef CAS PubMed; (b) J. Y. Jo, H. Y. Lee, W. J. Liu, A. Olasz, C. H. Chen and D. Lee, J. Am. Chem. Soc., 2012, 134, 16000 CrossRef CAS PubMed; (c) D. B. Zhao, Q. Wu, X. L. Huang, F. J. Song, T. Y. Lv and J. S. You, Chem. – Eur. J., 2013, 19, 6239 CrossRef CAS PubMed; (d) K. Muralirajan and C. H. Cheng, Chem. – Eur. J., 2013, 19, 6198 CrossRef CAS PubMed; (e) A. Alberti, N. Bedogni, M. Benaglia, R. Leardini, D. Nanni, G. F. Pedulli, A. Tundo and G. Zanardi, J. Org. Chem., 1992, 57, 607 CrossRef CAS; (f) X. L. Huang, X. Y. Chen and S. Ye, J. Org. Chem., 2009, 74, 7585 CrossRef CAS PubMed; (g) S. S. Mochalov, M. I. Khasanov, A. N. Fedotov and E. V. Trofimova, Chem. Heterocycl. Compd., 2008, 44, 229 CrossRef CAS PubMed.
- (a) T. Furuya, A. S. Kamlet and T. Ritter, Nature, 2011, 473, 470 CrossRef CAS PubMed.
- (a) H. J. Li, P. H. Li, Q. Zhao and L. Wang, Chem. Commun., 2013, 49, 9170 RSC; (b) H. Li, X. Y. Xie and L. Wang, Chem. Commun., 2014, 50, 4218 RSC.
- M. Sun, L. K. Hou, X. X. Chen, X. J. Yang, W. Sun and Y. S. Zang, Adv. Synth. Catal., 2014, 356, 3789 CrossRef CAS PubMed.
- M. L. Yi, X. L. Cui, C. W. Zhu, C. Pi, W. M. Zhu and Y. J. Wu, Asian J. Org. Chem., 2015, 4, 38 CrossRef CAS PubMed.
- (a) B. Xiao, Y. Fu, J. Xu, T. J. Gong, J. J. Dai, J. Yi and L. Liu, J. Am. Chem. Soc., 2010, 132, 468 CrossRef CAS PubMed; (b) B. Xiao, T. J. Gong, J. Xu, Z. J. Liu and L. Liu, J. Am. Chem. Soc., 2011, 133, 1466 CrossRef CAS PubMed; (c) R. Giri, J. K. Lam and J. Q. Yu, J. Am. Chem. Soc., 2010, 132, 686 CrossRef CAS PubMed.
- (a) X. Chen, K. M. Engle, D. H. Wang and J. Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094 CrossRef CAS PubMed; (b) L. M. Xu, B. J. Li, Z. Yang and Z. J. Shi, Chem. Soc. Rev., 2010, 39, 712 RSC; (c) K. M. Engle, T. S. Mei, M. Wasa and J. Q. Yu, Acc. Chem. Res., 2012, 45, 788 CrossRef CAS PubMed; (d) V. S. Thirunavukkarasu, S. I. Kozhushkov and L. Ackermann, Chem. Commun., 2014, 50, 29 RSC.
- (a) G. S. Georigiev, E. B. Kamenska, N. V. Tsarevsky and L. K. Christov, Polym. Int., 2001, 50, 313 CrossRef PubMed; (b) R. Amorati, L. Valgimigli, P. Dinér, K. Bakhtiari, M. Saeedi and L. Engman, Chem. – Eur. J., 2013, 19, 7510 CrossRef CAS PubMed.
- (a) Y. Siddaraju, M. Lamni and K. R. Prabhu, J. Org. Chem., 2014, 79, 3856 CrossRef CAS PubMed; (b) X. Mi, C. Y. Wang, M. M. Huang, Y. S. Wu and Y. J. Wu, J. Org. Chem., 2015, 80, 148 CrossRef CAS PubMed.
- (a) D. C. Powaers, M. A. L. Geibel, J. E. M. N. Klein and T. Ritter, J. Am. Chem. Soc., 2009, 131, 17050 CrossRef PubMed; (b) D. C. Powers and T. Ritter, Nat. Chem., 2009, 1, 302 CrossRef CAS PubMed; (c) J. M. Racowski, A. R. Dick and M. S. Sanford, J. Am. Chem. Soc., 2009, 131, 10974 CrossRef CAS PubMed; (d) N. R. Deprez and M. S. Sanford, J. Am. Chem. Soc., 2009, 131, 11234 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Preparation of azoxybenzene, the spectral data for all products and crystallographic data for 3q, 4l and complex I (CIF). CCDC 1030211, 1024177 and 1026842. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5qo00120j |
| This journal is © the Partner Organisations 2015 |