
Pd(II)-catalyzed intermolecular enantioselective hydroamination of styrenes†
Feng
Yu
,
Pinhong
Chen
* and
Guosheng
Liu
*
State Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, 345 Lingling Road, Shanghai, China. E-mail: pinhongchen@sioc.ac.cn; gliu@mail.sioc.ac.cn; Fax: +86-21-64166128; Tel: +86-21-54925346
First published on 7th May 2015
Abstract
A Pd-catalyzed intermolecular asymmetric hydroamination of styrenes was developed to give various chiral benzyl amides exclusively, in which the oxidation-stable pyridine-oxazoline was used as the chiral ligand to provide moderate to good enantioselectivities.
Hydroamination presents one of the most efficient strategies to build up alkylamines, and has received much attention.1 Among them, enantioselective hydroamination is much more attractive but challenging.2 Compared to the significant progress in the intramolecular hydroamination of alkenes by rare-earth metals,3 group 4 metals (Zr, Ti)4 and main-group metal (Li, Mg) complexes,5 the intermolecular enantioselective hydroamination reactions are less explored. For instance, Hartwig and coworkers developed a Pd-catalyzed intermolecular hydroamination of vinylarenes with arylamines in 2000, in which only two enantioselective examples were included using (R)-BINAP as the ligand.6 Further enantioselective hydroamination with modified reaction conditions was reported by Takahashi and Lin.7 Later, Shibata8 and Hartwig9 independently demonstrated that the enantioselective hydroamination of alkenes was achieved by using an Ir catalyst and chiral phosphine ligands. Very recently, the Buchwald10a and Miura10b groups found that a copper catalyst was used to catalyze the asymmetric hydroamination of alkenes to provide excellent enantioselectivities. Herein, we report our recent study on the Pd-catalyzed intermolecular asymmetric hydroamination of styrenes under oxidative reaction conditions, in which N-fluorobenzenesulfonimide (NFSI) was used as the nitrogen source as well as the oxidant, and pyridine-oxazoline (POX) type ligands were used as the chiral ligands to deliver good enatioselectivities.
In 2011, our group developed a palladium-catalyzed intermolecular hydroamination of styrenes.11 As shown in Scheme 1 (top), this reaction was initiated by alcohol oxidation with a Pd(II) catalyst to provide palladium hydride species. After migratory insertion of styrene into the palladium hydride species, the formed benzyl–Pd intermediate was oxidized by NFSI to provide the final hydroamination product, in which a Pd(IV) was proposed as a key intermediate for the C–N bond formation. It is notable that the ligand bathocuproine plays an important role for the successful catalytic cycle. Based on these results, we investigated the enantioselective hydroamination reaction by employing chiral ligands. Compared to the chiral phosphine ligands used in the previous studies, nitrogen-based ligands, such as POX type ligands L1–L6, are more compatible with the oxidative reaction conditions, which have been widely used in the palladium-catalyzed oxidative transformations, including palladium-catalyzed intramolecular enantioselective oxidative amination of alkenes, asymmetric Heck reaction and others.12 In order to assess the potential of asymmetric hydroamination, these ligands were used in the initial study. As shown in Scheme 1 (bottom), we were delighted to find that the POX type ligand was effective toward hydroamination. When simple POX ligand L1 was used, the reaction did give the desired product 2a in 70% yield, but unfortunately without enantioselectivity. Very interestingly, the sterically bulkier ligand L2 bearing an ortho-methyl group on the pyridyl ring exhibited moderate enantioselectivity (43% ee) in good yield. Then a variety of POX ligands L3–L6 with different groups at the oxazoline ring were screened and L6 with the tert-butyl group provided the best result in 50% ee. After further optimization of the reaction conditions, the ee value of 2a could be increased to 60% in EtOAc.
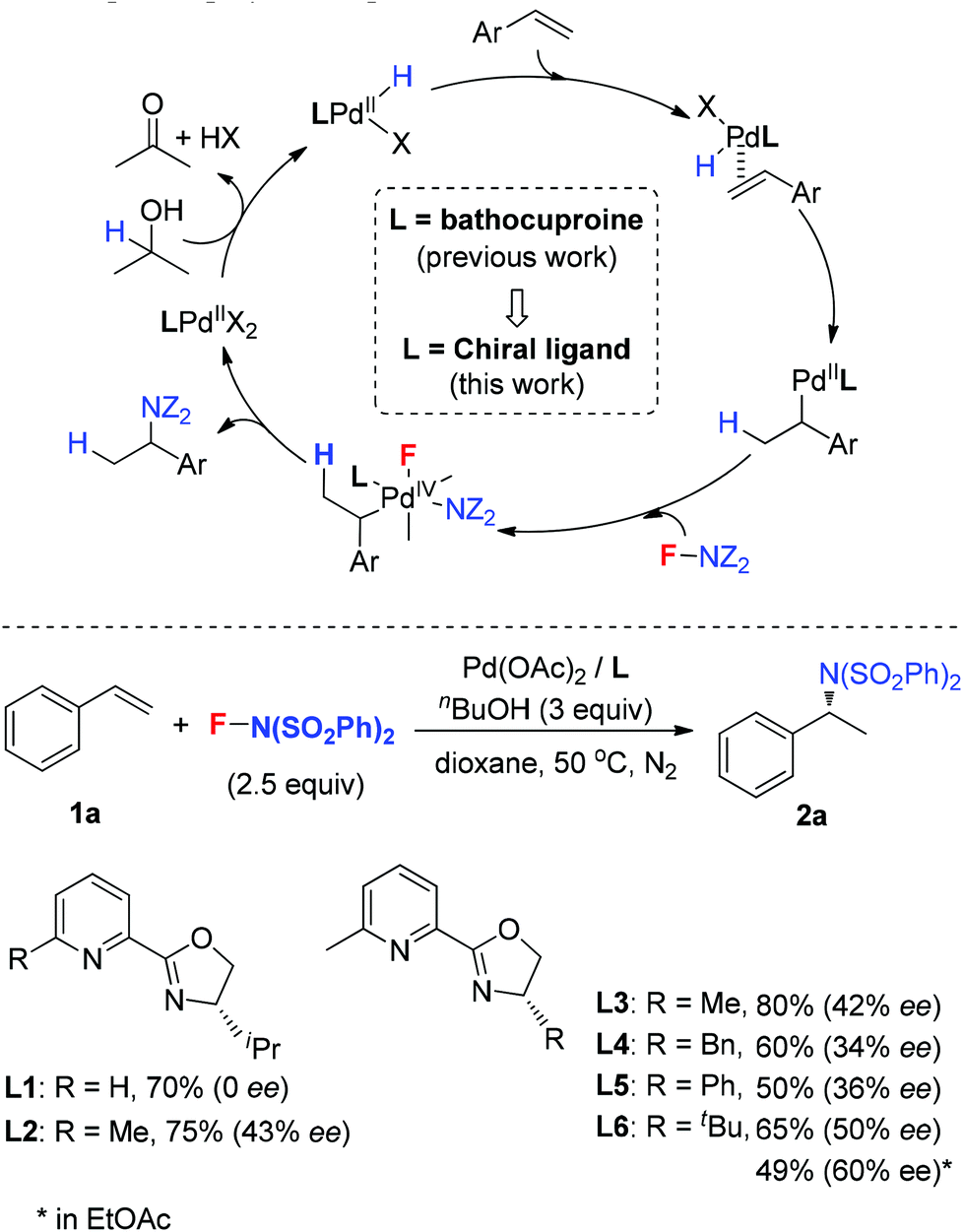 | ||
| Scheme 1 Pd(II)-catalyzed asymmetric hydroamination of styrene. | ||
Our previous study revealed that the reductive elimination of the Pd(IV) intermediate undergoes an exclusive SN2 type pathway.13 Thus, the enantioselective migratory insertion step is responsible for the final ee value of the product. We reasoned that enlargement of the steric hindrance of ligands might be helpful to increase the enantioselectivity of migratory insertion. In order to test the above hypothesis, a series of POX ligands bearing different ortho-substituents on the pyridyl ring (L7–L18) were further synthesized and employed in the reaction (Fig. 1). As shown in Table 1, when L7–L9 with Et, nPr and iPr groups were used to catalyze the reaction, slightly better enantioselectivities were obtained (entries 1–3). However, L10 and L11 with Ph and Bn groups exhibited poor reactivity (entries 4 and 5). Compared with the above ligands, L12 with the iBu group presented the best enantioselectivity to afford 2a in 79% ee but with low yield (36%, entry 6). The reaction yield could be improved to 63% by increasing the catalyst loading to 10 mol% with a similar ee value (entry 7). Furthermore, the addition of triethyl orthoformate was beneficial to give a better reaction yield as well as enantioselectivity (entry 8). With these modified reaction conditions, extra ligands L13–L18 were further screened. These ligands exhibited slightly poor selectivities compared to L12 (entries 9–14). Finally, the best result (87% ee and 75% yield) was obtained by increasing the amount of NFSI (3 equiv., entry 15). It should be noted here that the chiral amine 2a was determined to have the (S)-configuration according to the X-ray structure (Fig. 2).
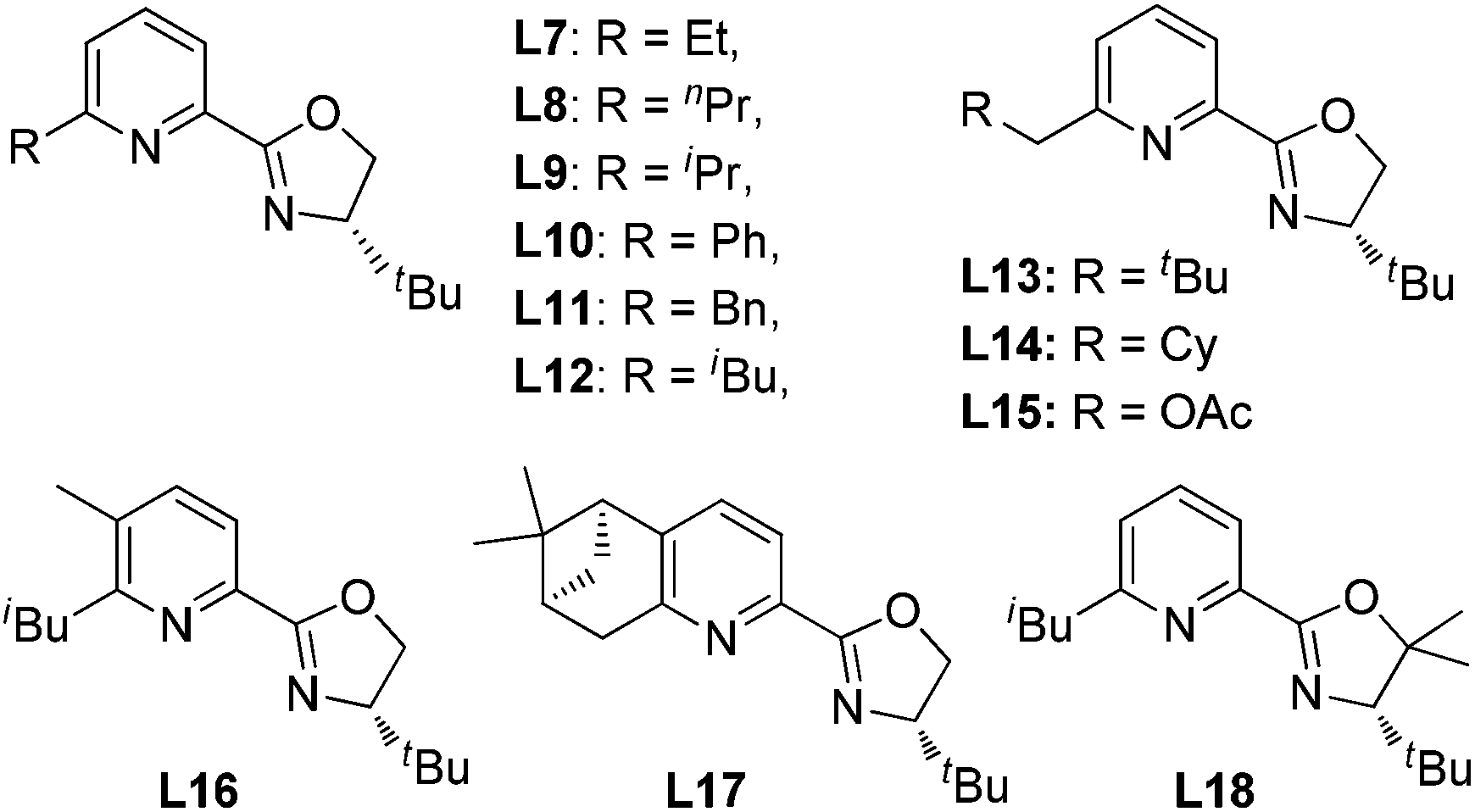 | ||
| Fig. 1 The structure of POX ligands. | ||
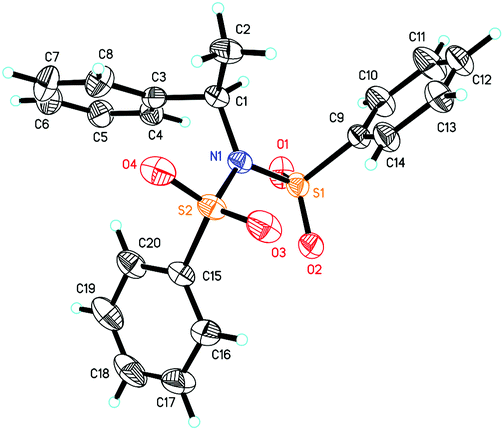 | ||
| Fig. 2 The X-ray structure of 2a. | ||
|
|
|||
|---|---|---|---|
| Entry | Ligand | 2a Yields (%) | ee (%) |
| a Conditions: 1a (0.2 mmol), Pd(OAc)2 (5 mol%), L (7.5 mol%), NFSI (2.5 equiv.) in EtOAc, at 30 °C. b Pd(OAc)2 (10 mol%), ligand (15 mol%). c HC(OEt)3 (1.5 equiv.). d NFSI (3.0 equiv.). | |||
| 1 | L7 | 57 | 73 |
| 2 | L8 | 46 | 77 |
| 3 | L9 | 50 | 65 |
| 4 | L10 | Trace | — |
| 5 | L11 | 16 | 49 |
| 6 | L12 | 36 | 79 |
| 7b | L12 | 63 | 82 |
| 8b,c | L12 | 72 | 86 |
| 9b,c | L13 | 40 | 71 |
| 10b,c | L14 | 75 | 80 |
| 11b,c | L15 | 45 | 59 |
| 12b,c | L16 | 40 | 75 |
| 13b,c | L17 | 52 | 79 |
| 14b,c | L18 | 50 | 75 |
| 15b,c,d | L12 | 75 | 87 |
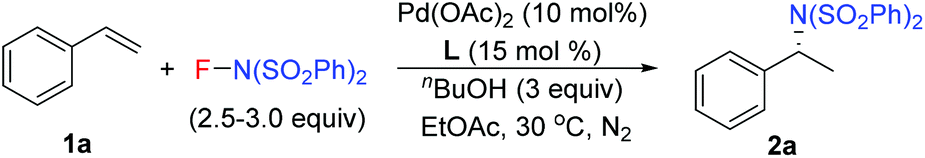
With the optimized reaction conditions in hand, the scope of styrene was subsequently examined. As shown in Table 2, the styrenes with halides at the para position afforded the corresponding chiral amides 2b–2e in good ee values and yields. For the product 2e, L6 with small steric hindrance provided better yield and poor enantioselectivity than that of L12. Compared to the electron-deficient halide group, the electron-rich styrenes gave slightly lower ee values to generate products 2f–2h. For the meta-substituted styrenes, good enantioselectivity was also obtained to generate products 2i–2k but with poor yields, especially for the cases of substrates with a meta-halide. However, these substrates reacted smoothly under the reaction conditions with L6 as the ligand to afford the desired products 2j–2l in good yields and ee values. Furthermore, the current reaction conditions were also compatible for the ortho-methyl styrene and dimethyl styrene to give products 2m–2n in moderate yields and ee values. However, ortho-halide substituted styrenes were ineffective. It is notable that, unfortunately, styrenes with a strong electron-withdrawing group, such as ester, nitro, and ketone, were ineffective for the transformation.
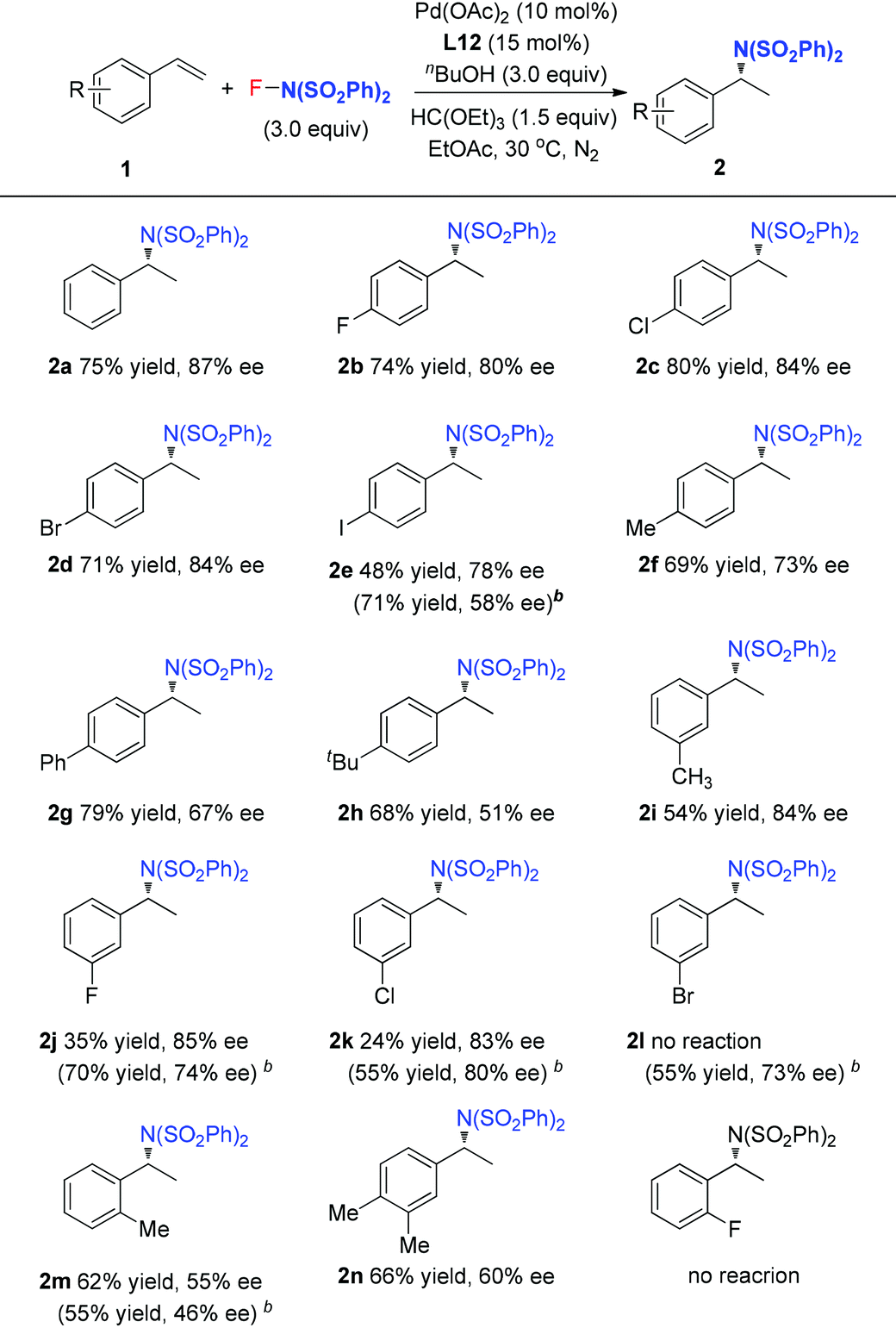
The above observations revealed that, compared to L6, the large steric hindrance of ligand L12 exhibited a negative impact on the reactivity but a positive impact on the enantioselectivity. Originally, we reasoned that ligand L12 bearing two substituents in both oxazoline and pyridine might act as a pseudo C2-symmetric ligand to coordinate with palladium(II) in the reaction system (Fig. 3A), which is helpful to control the stereochemistry of hydropalladation. Surprisingly, the X-ray of complex (L12)PdCl2 showed the two substituents in the same face (Fig. 3B). In addition, the X-ray structure indicated that there are two (L12)PdCl2 in a unit lattice, and a weak interaction between the two palladium centers is observed (Pd(1)–Pd(2) = 3.854 Å), and the two ligands are aligned parallel, which forces the two substituents in the same face to lower the lattice energy, due to the flexible iBu group (see ESI†). These observations reveal that the rotation of the iPr group of the (L12)PdCl2 complex should be easier than our expectation.
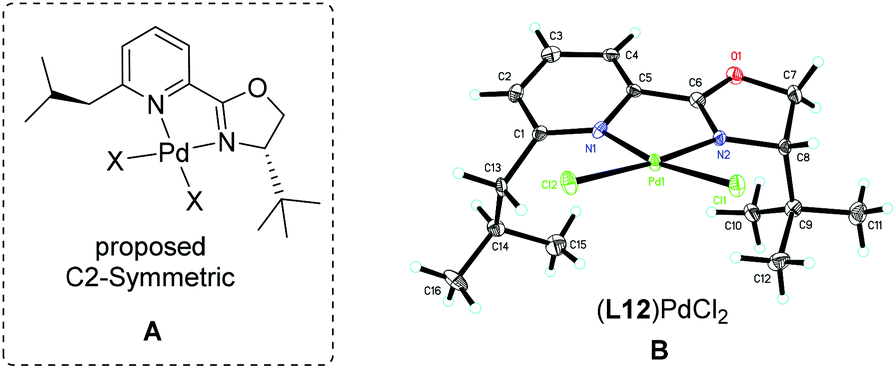 | ||
| Fig. 3 The X-ray structure of complex (L12)PdCl2. | ||
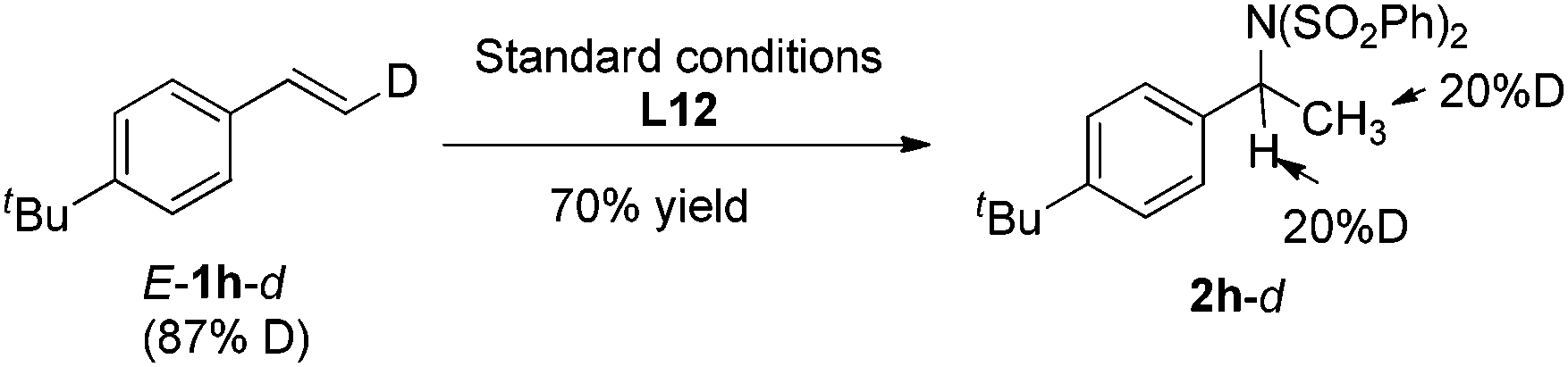
Furthermore, deuterium labeled styrene E-1h-d was subjected to the standard conditions, and the hydroamination product 2h-d was obtained as a mixture of isomers with the deuterium incorporation on both α and β carbon (eqn (1)). In addition, when substrate 1h was treated under the standard conditions with iPrOH-d8, the same product 2h-d was obtained (eqn (2)). These observations indicated that a quick reversible migratory insertion of styrene into the palladium hydride species was involved in the reaction, which is possible to ruin the enantioselectivity to some degree.
 | (1) |
 | (2) |
In conclusion, we have described a Pd-catalyzed intermolecular enantioselective hydroamination of styrenes, which afforded various chiral branched amides with excellent regio- and good enantioselectivity, in which the modified pyridine-oxazoline ligand was used under oxidation conditions. Preliminary mechanistic understanding reveals that the modified ligand presented a good enantioselective control on the migratory insertion step. However, the ligand structure still needs to be improved to act as a pseudo C2-symmetric ligand in future study. In addition, we hope this new type of ligand could also improve the sequential process of benzyl C–Pd(II) oxidation to C–Pd(IV) and the following reductive elimination, which could reduce the β-H elimination of the benzyl C–Pd(II). The related investigation is in progress in our laboratory.
We are grateful for financial support from the National Basic Research Program of China (973-2015CB856600), the National Nature Science Foundation of China (no. 21225210, 21421091, and 21472217), and the Science Technology Commission of the Shanghai Municipality (12ZR1453400). P.H.C. also thanks the Key Laboratory of Functional Small Organic Molecules, Ministry of Education, Jiangxi Normal University (no. KLFS-KF-201402), for the financial support.
Notes and references
- For some reviews on the hydroamination: (a) T. E. Müller and M. Beller, Chem. Rev., 1998, 98, 675 CrossRef PubMed; (b) M. Norbis and B. D. Hölscher, Angew. Chem., Int. Ed., 2001, 40, 3983 CrossRef; (c) N. Nishina and Y. Yamamoto, Top. Organomet. Chem., 2013, 43, 115 CrossRef CAS; (d) E. Chong, P. Garcia and L. L. Schafer, Synthesis, 2014, 2884 Search PubMed; (e) A. L. Reznichenko, A. J. Nawara-Hultzsch and K. C. Hultzsch, Top. Curr. Chem., 2014, 343, 191 CrossRef; (f) L. Huang, M. Arndt, K. Gooßen, H. Heydt and L. J. Gooßen, Chem. Rev., 2015, 115, 2596 CrossRef CAS PubMed.
- For the reviews on the asymmetric hydroamination, see: (a) K. C. Hultzsch, Adv. Synth. Catal., 2005, 347, 367 CrossRef CAS PubMed; (b) H. Jerome and S. Emmanuelle, Chem. – Eur. J., 2013, 19, 4972 CrossRef PubMed; (c) G. Zi, J. Organomet. Chem., 2010, 696, 68 CrossRef PubMed; (d) K. D. Hesp, Angew. Chem., Int. Ed., 2014, 53, 2034 CrossRef CAS PubMed; (e) P. W. Roesky and T. E. Mueller, Angew. Chem., Int. Ed., 2003, 42, 2708 CrossRef CAS PubMed.
- (a) M. R. Gagne, L. Brard, V. P. Conticello, M. A. Giardello, C. L. Stern and T. J. Marks, Organometallics, 1992, 11, 2003 CrossRef CAS; (b) S. Hong, A. M. Kawaoka and T. J. Marks, J. Am. Chem. Soc., 2003, 125, 15878 CrossRef CAS PubMed; (c) Y. Chapurina, J. Hannedouche, J. Collin, R. Guillot, E. Schulz and A. Trifonov, Chem. Commun., 2010, 46, 6918 RSC; (d) Y. Zhang, W. Yao, H. Li and Y. Mu, Organometallics, 2012, 31, 4670 CrossRef CAS; (e) A. L. Reznichenko, H. N. Nguyen and K. C. Hultzsch, Angew. Chem., Int. Ed., 2010, 49, 8984 CrossRef CAS PubMed.
- (a) D. A. Watson, M. Chiu and R. G. Bergman, Organometallics, 2006, 25, 4731 CrossRef CAS PubMed; (b) A. L. Gott, A. J. Clarke, G. J. Clarkson and P. Scott, Organometallics, 2007, 26, 1729 CrossRef CAS; (c) M. C. Wood, D. C. Leitch, C. S. Yeung, J. A. Kozak and L. L. Schafer, Angew. Chem., Int. Ed., 2006, 46, 354 CrossRef PubMed; (d) K. Manna, S. Xu and A. D. Sadow, Angew. Chem., Int. Ed., 2011, 50, 1865 CrossRef CAS PubMed.
- (a) P. Horrillo Martínez, K. C. Hultzsch and F. Hampel, Chem. Commun., 2006, 2221 RSC; (b) J. Deschamp, C. Olier, E. Schulz, R. Guillot, J. Hannedouche and J. Collin, Adv. Synth. Catal., 2010, 352, 2171 CrossRef CAS PubMed; (c) X. Zhang, T. J. Emge and K. C. Hultzsch, Angew. Chem., Int. Ed., 2012, 51, 394 CrossRef CAS PubMed.
- (a) M. Kawatsura and J. F. Hartwig, J. Am. Chem. Soc., 2000, 122, 9546 CrossRef CAS; (b) U. Nettekoven and J. F. Hartwig, J. Am. Chem. Soc., 2002, 124, 1166 CrossRef CAS PubMed; (c) M. Utsunomiya and J. F. Hartwig, J. Am. Chem. Soc., 2003, 125, 14286 CrossRef CAS PubMed.
- A. Hu, M. Ogasawara, T. Sakamoto, A. Okada, K. Nakajima, T. Takahashi and W. Lin, Adv. Synth. Catal., 2006, 348, 2051 CrossRef CAS PubMed.
- S. Pan, K. Endo and T. Shibata, Org. Lett., 2012, 14, 780 CrossRef CAS PubMed.
- (a) C. S. Sevov, J. Zhou and J. F. Hartwig, J. Am. Chem. Soc., 2012, 134, 11960 CrossRef CAS PubMed; (b) C. S. Sevov, J. Zhou and J. F. Hartwig, J. Am. Chem. Soc., 2014, 136, 3200 CrossRef CAS PubMed.
- (a) S. Zhu, N. Niljianskul and S. L. Buchwald, J. Am. Chem. Soc., 2013, 135, 15746 CrossRef CAS PubMed; (b) Y. Miki, K. Hirano, T. Satoh and M. Miura, Angew. Chem., Int. Ed., 2013, 52, 10830 CrossRef CAS PubMed.
- T. Xu, S. Qiu and G. Liu, J. Organomet. Chem., 2011, 696, 46 CrossRef CAS PubMed.
- For some recent examples, see: (a) A. B. Weinstein and S. S. Stahl, Angew. Chem., Int. Ed., 2012, 51, 11505 CrossRef CAS PubMed; (b) G. Yang, C. Shen and W. Zhang, Angew. Chem., Int. Ed., 2012, 51, 9141 CrossRef CAS PubMed; (c) J. C. Holder, L. Zou, A. N. Marziale, P. Liu, Y. Lan, M. Gatti, K. Kikushima, K. N. Houk and B. M. Stoltz, J. Am. Chem. Soc., 2013, 135, 14996 CrossRef CAS PubMed; (d) T.-S. Mei, E. W. Werner, A. J. Burckle and M. S. Sigman, J. Am. Chem. Soc., 2013, 135, 6830 CrossRef CAS PubMed; (e) Y. Zhang and M. S. Sigman, J. Am. Chem. Soc., 2007, 129, 3076 CrossRef CAS PubMed; (f) E. L. Ingalls, P. A. Sibbald, W. Kaminsky and F. E. Michael, J. Am. Chem. Soc., 2013, 135, 8854 CrossRef CAS PubMed; (g) W. He, K.-T. Yip, N.-Y. Zhu and D. Yang, Org. Lett., 2009, 11, 5626 CrossRef CAS PubMed.
- H. Peng, Z. Yuan, H.-Y. Wang, Y.-L. Guo and G. Liu, Chem. Sci., 2013, 4, 317 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1055926. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5qo00096c |
| This journal is © the Partner Organisations 2015 |